
Pharmacology Study Notes
药理学绪论
药理学概念
什么是药物?
- 药物概念:用于防治、诊断疾病和计划生育的化学物质
- 药物和毒物只有剂量上的差异
- 思维方式:从基本作用点推理系统变化
- 药物命名:国际通用名、商品名、化学名
- 命名规律:
如他汀类药物:降低胆固醇药——羟甲基二酰辅酶A(HMG-CoA)还原酶(胆固醇合成通路中的限速酶)抑制剂 - 药物分类:1.按化学分类 2.按作用机理分类 3.按治疗疾病分类
什么是药理学
- 概念:药理学是研究药物与机体之间相互作用规律的一门科学。药效学(Pharmacodynamics)是研究药物对机体的作用或在药物影响下机体细胞功能如何发生变化;药物代谢动力学(pharmacokinetics)简称药代动力学或药动学,主要是定量研究药物在生物体内的过程(吸收、分布、代谢和排泄),并运用数学原理和方法阐述药物在机体内的动态规律的一门学科。adme/t
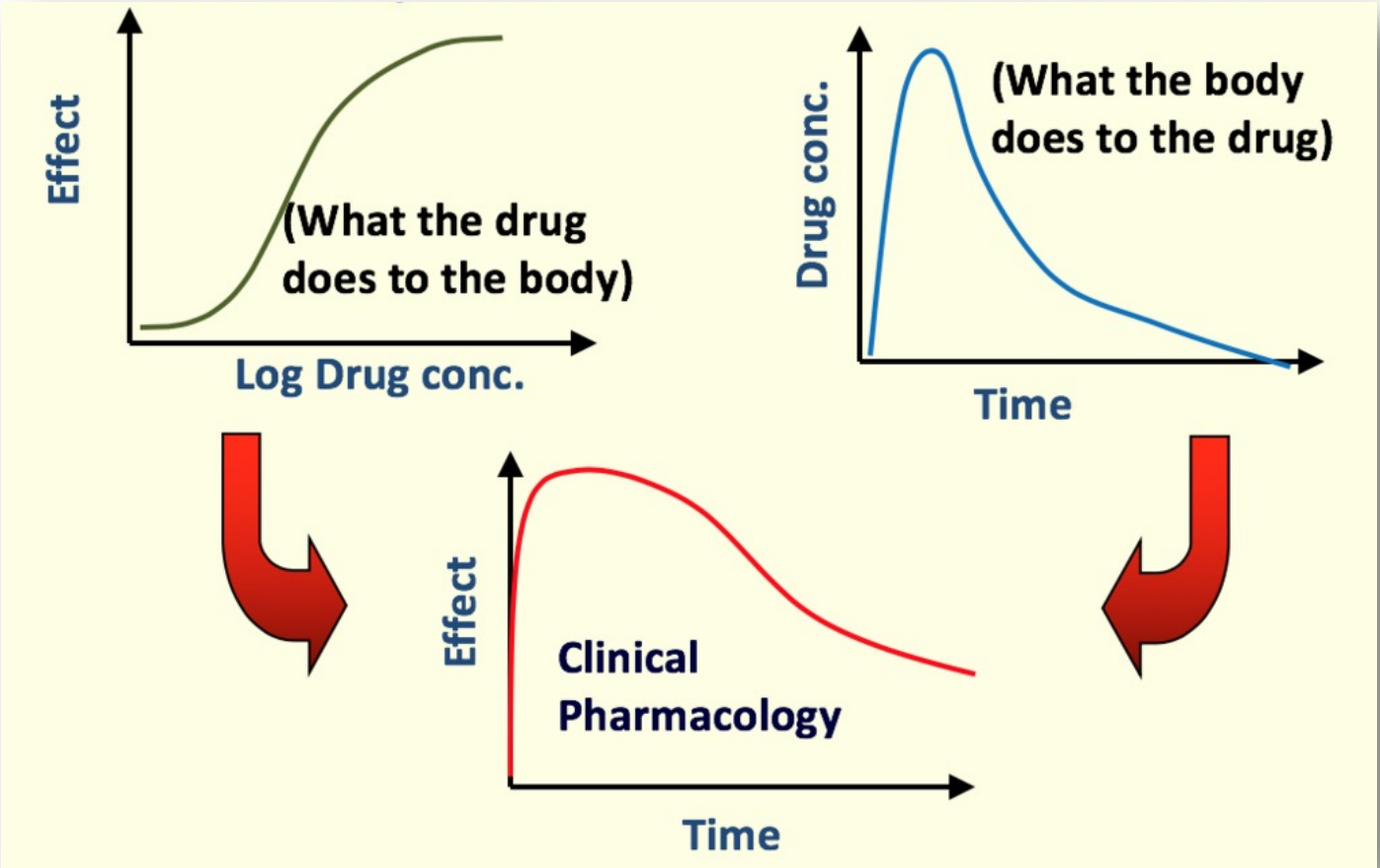
- What the drug does to the body: pharmacodynamics 药效学
- What the body does to the drug: pharmacokinetics 药动学
- 药理效应:药物分子必须对细胞的一个或多个成分产生化学作用
- 药物靶点:药物分子和特定组织细胞成分结合的区域
- 药效学(治疗作用+不良反应):
- 药物的适应症(indications)
- 药物作用机理(mechanism)
- 药物的不良反应(ADRs)
- 药物的禁忌症(contraindications)
- 药动学(ADME)
- 药物如何吸收(Absorption)
- 药物如何分布 (Distribution)
- 药物如何代谢(Metabolism)
- 药物如何排泄(Elimination)
- 了解如何用药达到治疗窗口
- 概念词:in vitro 离体; in vivo 在体; (ex vivo) 离体再入体; in situ 原位; Pre-clinical study 临床前研究, clinical trial 临床试验
现代新药研发
- 现代生物制药的原点:外部活性成分能够作用于特定靶点,是一定存在内源性的配体相结合
- Reverse pharmacology: Find medicinable target -> new chemicals -> drugs(成功率更高)
- 现代新药研发过程:Proof of Mechanism; Proof of Concept; Proof of Principle
药代动力学(Pharmacokinetics)
药物代谢动力学概念
-
概念: 研究药物体内过程以及体内药物浓度随时间变化的规律,身体如何代谢影响药物
-
药代动力学研究范围:
-
药物吸收模型:
药物的转运方式
- 跨膜转运
- 被动转运
- 简单扩散
- 脂溶扩散(脂/水分配系数、浓度梯度、膜面积、膜厚度、解离度)
- 水溶扩散(直径小于膜孔)
- 易化扩散(不耗能、需载体)
- 简单扩散
- 主动转运(耗能、要载体)
- 原发性
- 继发性
- 交换转运
- 协同转运
- 膜动转运(耗能不需载体)
- 胞吞
- 胞吐
- 被动转运
a.脂溶扩散
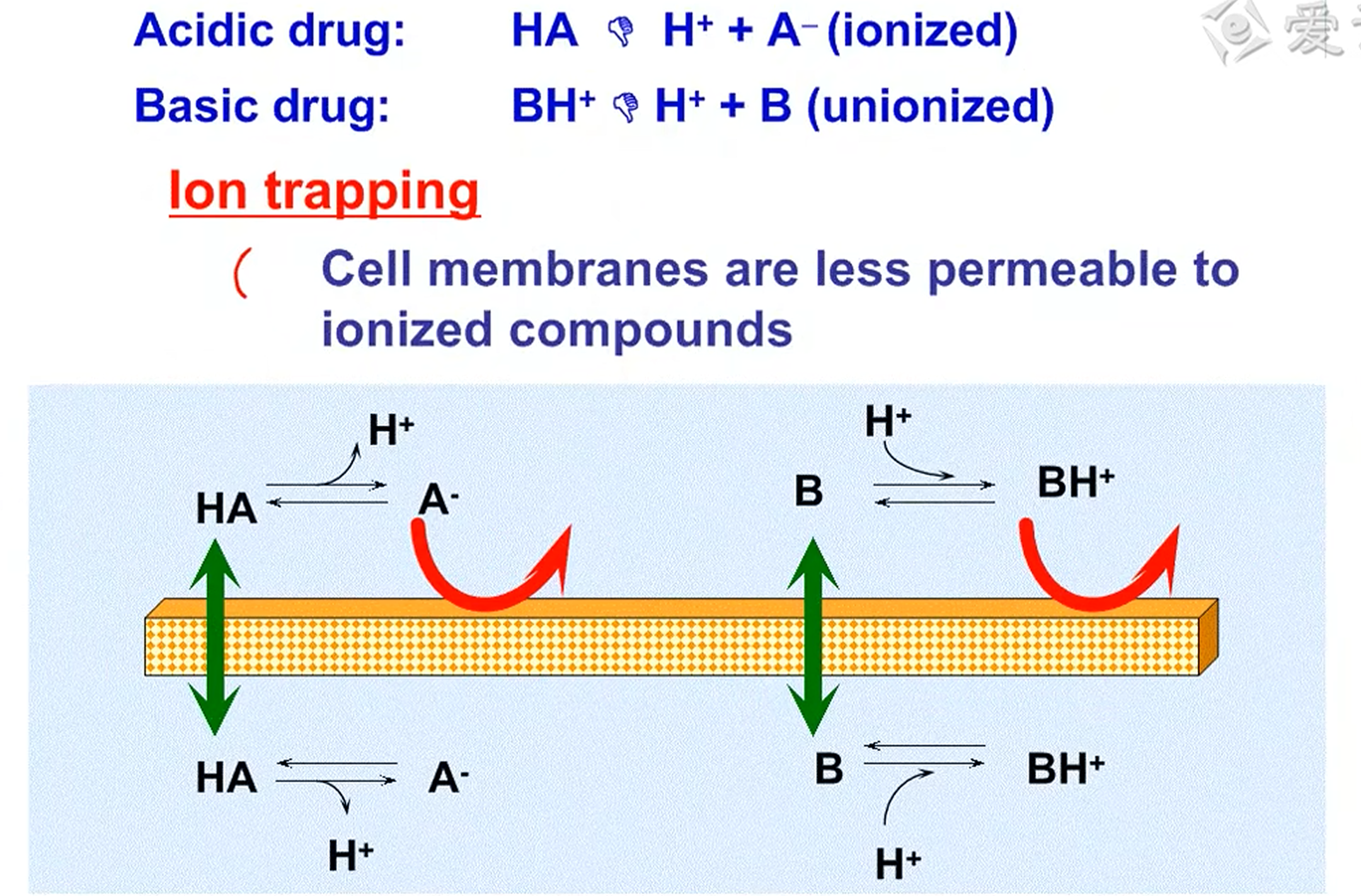
- 脂溶扩散:属于passive transport,顺浓度梯度、不耗能、无载体饱和性、无竞争性(大多数药物的转运方式)
- 离子障(ion trapping):离子型型药物被限制在一侧,非离子型药物会自由穿透
- 因此pH值会很大程度上影响药物的吸收:pH改变了药物的带电荷数目,影响其离子或非离子形态
绝大多数药物一般都具有脂溶性,大都是以脂溶扩散方式进入细胞内,因为很难有特定的运输载体(人造分子),且都从给药部位扩散到靶点
大多数药物的吸收部位在肠道,因为肠道的面积大,而不是在很多看上去吸收快的地方(比如胃可能吸收快,但是面积小)
举例:苯巴比妥(弱酸性)pKa=7.4,根据 ,当尿液为碱性时,pH值大于pKa, 增多,即解离型多,重吸收减少,药物排泄加快,所以中毒时应该碱化尿液————这是促进弱酸性药物加快通过肾脏排泄的通用办法
b.水溶扩散
水溶扩散:水溶性小分子通过水孔蛋白进行扩散(仅小分子:水酒精尿素,分子量不能超过100)
该种方式在肌肉注射剂中使用多,因为细胞间隙水溶性物质扩散更快,进一步原因是血管和内皮之间间隙远大于细胞膜,注射药物更容易在组织液和血浆之间扩散
分子量较小的通过毛细血管吸收,更大的可以通过毛细淋巴管吸收
c.易化扩散
- 药物转运体(transporter)
- 摄取性转运体:PEPT1(水溶性 内酰类口服更易吸收)
- 外排性转运体: P-gp(p-糖蛋白)————耐药基因
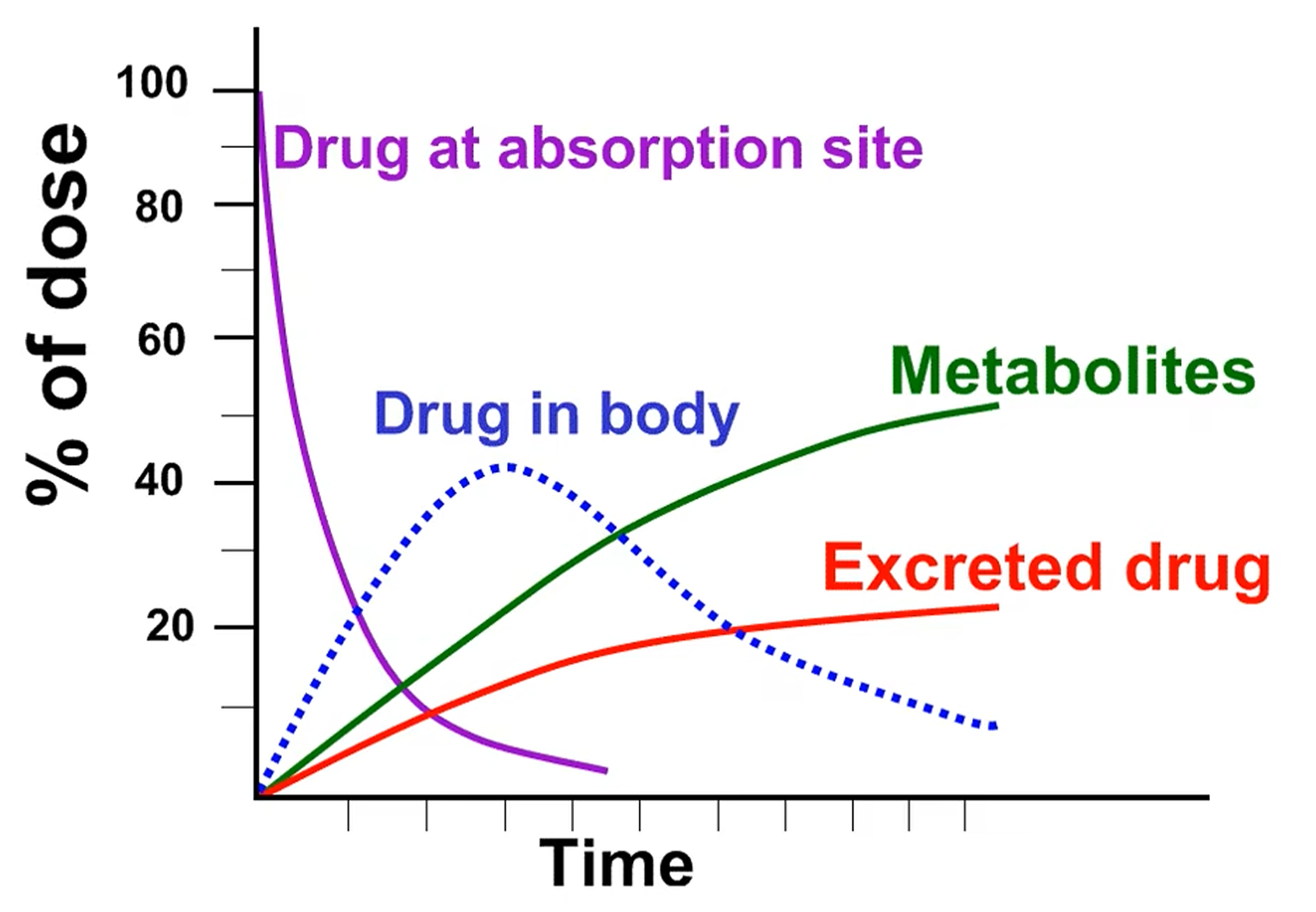
药物体内代谢过程 ADME (Adsorption, Distribution,Metabolism, Excretion)
a. Adsorption
-
吸收(Adsorption): Transfer of a drug from its site of administration to the bloodstream
-
Movement of a drug across cell barrier
-
Diffusion through lipid(Non-polar)
- Meaning: Let drug can easily across the cell membrane only by diffusion
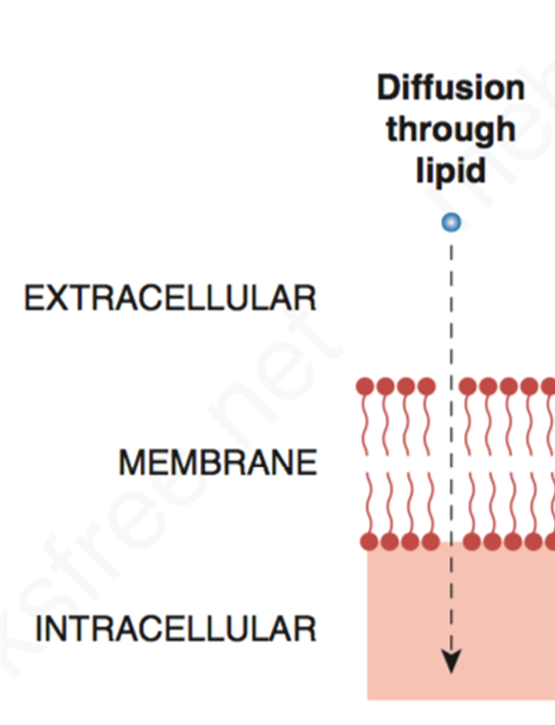
- Meaning: Let drug can easily across the cell membrane only by diffusion
-
ion trapping
- Meaning: Bring polar to drugs which can help drug to stay in a certain place of cell. If a drug is ionized, it can not across membrane or move to other body compartment. Each compartment of cell has different pH so we can modify drug to keep it stay where we want.
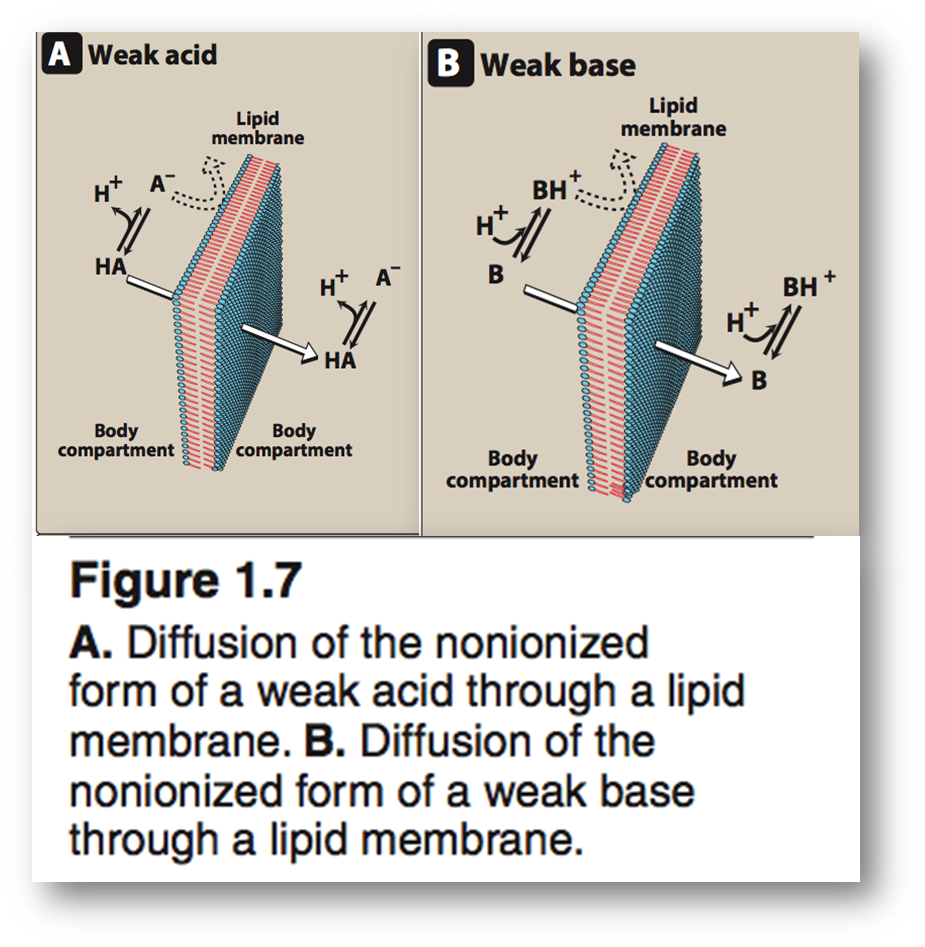
- Meaning: Bring polar to drugs which can help drug to stay in a certain place of cell. If a drug is ionized, it can not across membrane or move to other body compartment. Each compartment of cell has different pH so we can modify drug to keep it stay where we want.
-
Carrier-mediated transport
- Meaning: Drug molecules may be transport by OCT family into liver and take metabolism and excretion process
Carrier has genetic variation and are targets for drug interactions
- SLCs:solute carriers
- ATP-binding cassette [ABC] transporters, also referred to as “multi-drug resistance protein”
- Organic cation transporters (OCTs), and organic anion transporters (OATs)
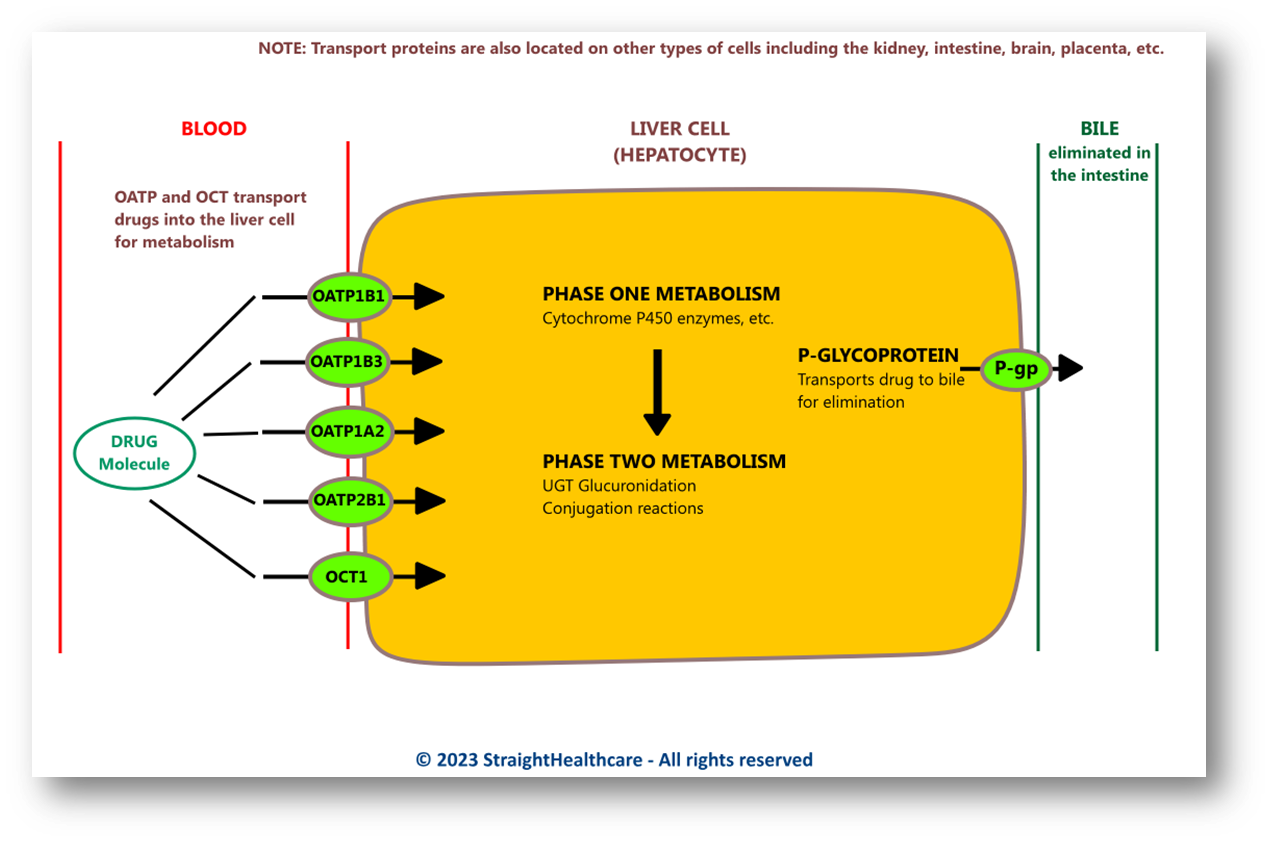
-
Endocytosis and exocytosis
- Most large molecules use this pathway by using vesicle system, like Vitamin B12, antibody or neurotransmitters
-
-
Routes of administration
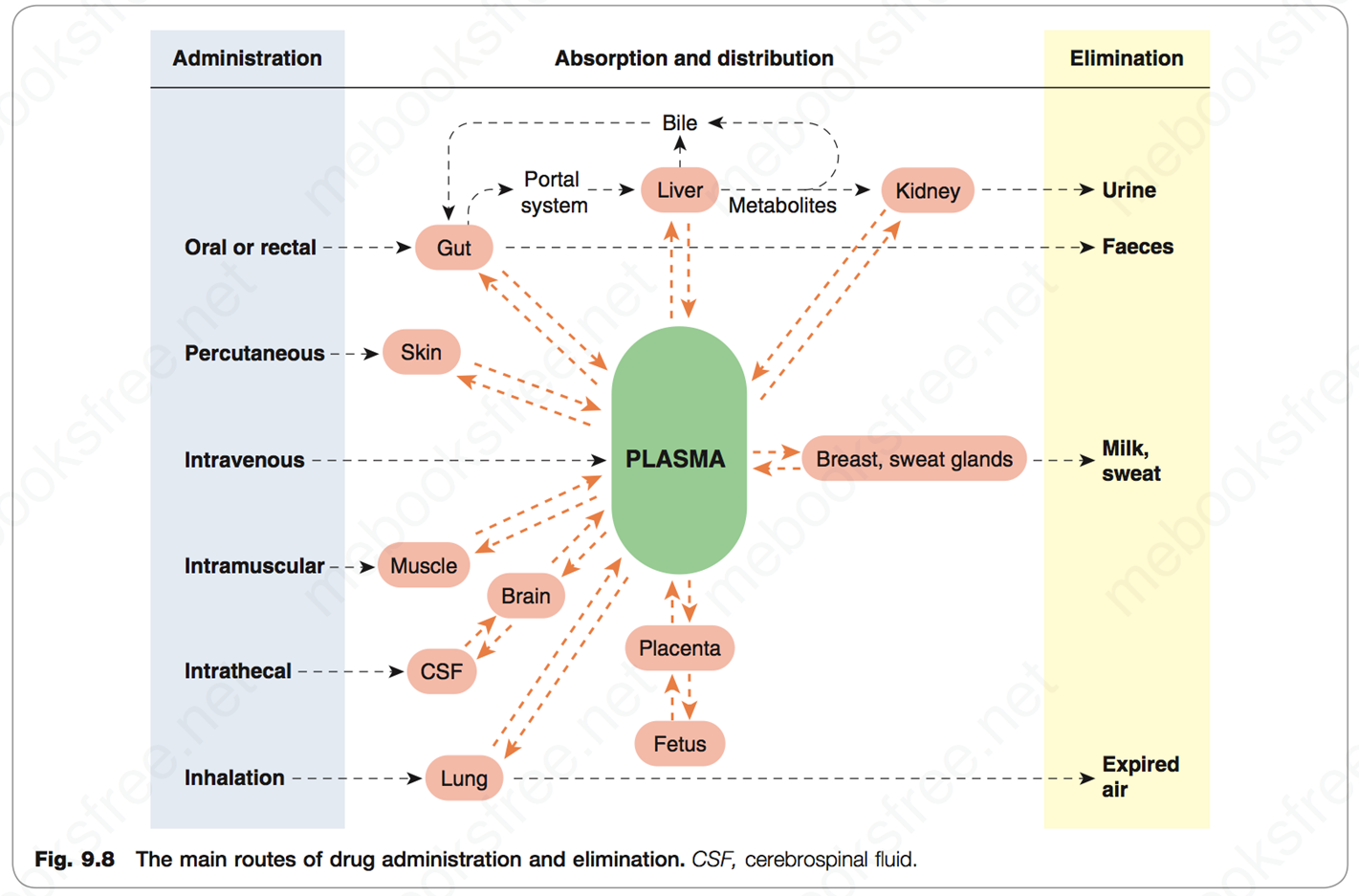
-
口服给药(Oral ingestion)
- 主要作用部位(Major site): intestine 肠道
- 平均转移时间长:可以达到3h
- 非常大的肠道表面积(有大量微绒毛)
- 血流量丰富,毛细血管众多
- 优劣比较
- 方便,剂量相对安全可控,成本低
- 吸收面积很大,无疼痛
- ph5-8适合大多数药物的吸收
- 达峰时间长,不适用紧急情况
- 不能用于无意识患者
- 首过效应(First Pass Effect)
- 定义:药物分子经过口服给药在初次进入体内,没有进入血液循环,而在肠道和肝脏被代谢,使得进入血液循环原药量减少的现象————造成吸收不全的主要原因
- 首过效应过强的药物不适合肠道给药,如:硝酸甘油(95%,一般选取舌下含化,或者贴软膏依靠透皮吸收)
- 主要作用部位(Major site): intestine 肠道
-
静脉注射(Intravenous)
- 主要作用部位:静脉
- 药物作用迅速,快速达到峰值浓度
- 剂量不易控制,达峰快容易引起中毒
- 主要作用部位:静脉
-
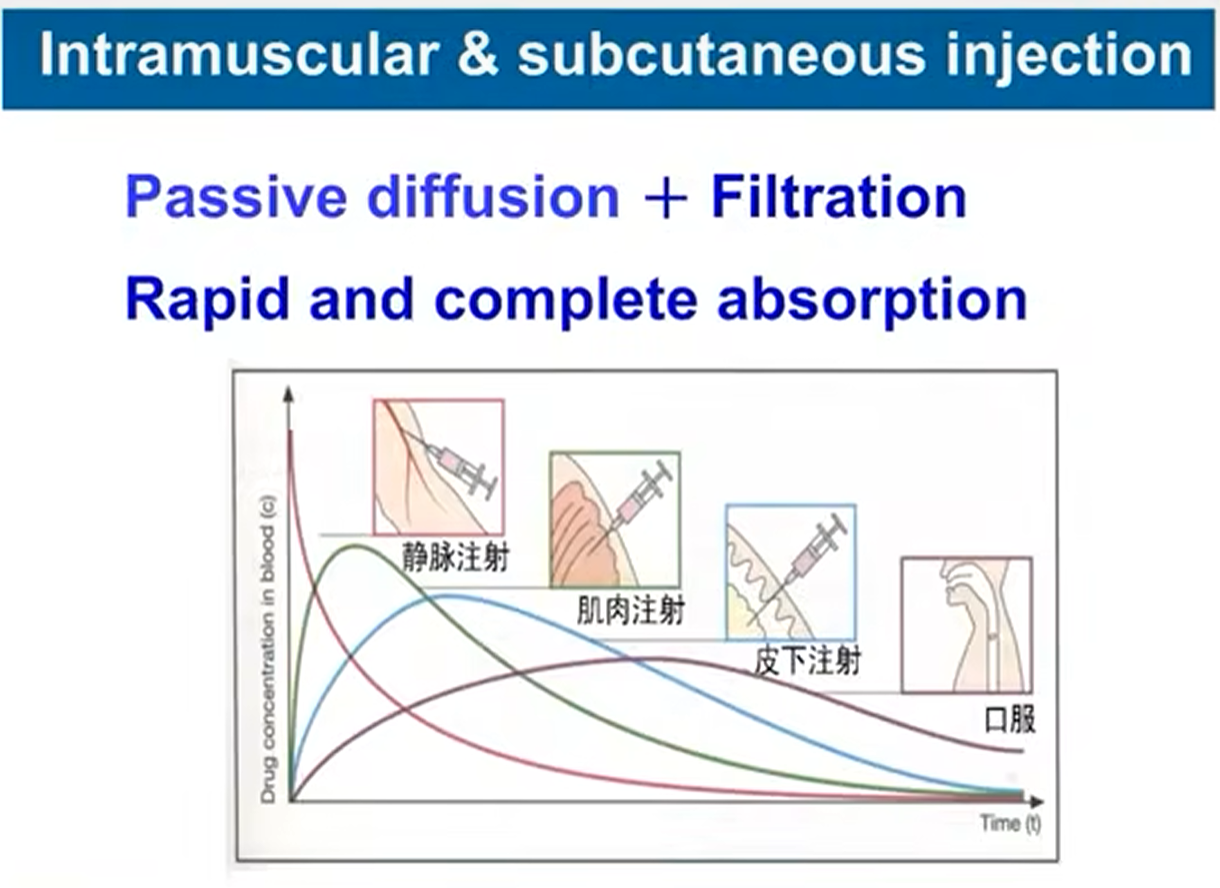
肌肉注射/皮下注射(Intramuscular & subcutaneous injection )
- 主要作用部位:肌肉
- 容易控制吸收速度和剂量
- 疼痛,不方便
- 主要作用部位:肌肉
-
脑脊液注射/玻璃体注射(Intrathecal & intravitreal)
-
吸入给药(Inhalation)
- 主要作用部位:lung 肺部
- 平均转移时间短,没有首过效应,直接到达作用部位
- 肺泡的表面积非常大
- 主要作用部位:lung 肺部
-
口腔粘膜给药
- 主要作用部位: nasal 鼻, oral 口, buccal 颊
- 快速吸收,剂量低
- 可以避免首过效应
- 不方便,药物有异味
- 主要作用部位: nasal 鼻, oral 口, buccal 颊
-
透皮吸收(Transdermal)
- 主要作用部位: skin 皮肤 mucous membranes 粘膜
- 必须为脂溶性药物,需要很强的脂溶性
- 吸收速度非常慢,剂量易控
- 主要作用部位: skin 皮肤 mucous membranes 粘膜
几种给药方式的药物浓度变化曲线:
b. Distribution
-
分布(distribution):how a drug reaches its target organ
-
主要影响因素

- 理化特性(脂溶性、离子化)、分子大小
- 心脏收缩量、组织冠流量
- 毛细血管通透性(注意blood brain barrier)
- 组织脂肪含量
- 血浆蛋白结合率
- 硫喷妥钠:超短作用的巴比妥类药物,常用于短时间内麻醉,主要为静脉注射给药。
- 特点:
1.超速效:极强的脂溶性(水油分配系数很高),静脉注射后会非常快通过血液循环进入大脑组织(快于进入脂肪组织),进入脑细胞。抑制中枢神经造成意识丧失
2.超短效:由于极强脂溶性,脑内高浓度药物也会随血液循环快速转移至脂肪组织(药物和脂肪亲和力最大,能溶解大量药物),从而脑中浓度下降,意识恢复
+ 作用:诱导麻醉药物
+ 多次给药特点: “VUL”充分说明意识恢复并不代表药物的降解排泄,而是药物再分配到其他部位,多次给药体内累计药物浓度达到致死剂量造成死亡
再分配(redistribution): 药物从第一作用位点重新分布到第二作用位点,从而使得第一位点的药物浓度下降,而非由于药物降解排泄导致。
-
血浆蛋白结合率
- 血浆蛋白结合特点
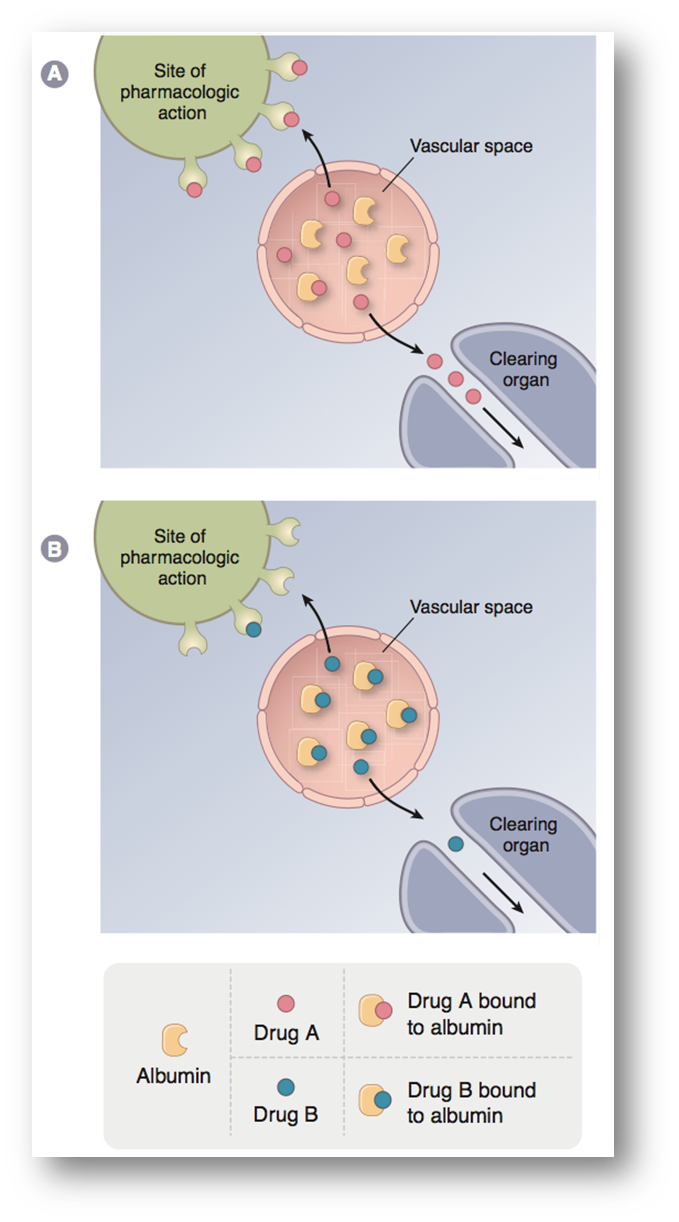
- 可逆,存在动态平衡
- 可饱和,结合位点有限
- 存在竞争性,非特异性结合
- 使结合的药物暂时失去活性(限制药物扩散到血管之外,由于可逆结合,低浓度时释放,以达到缓释效果)
- 结合规律
- 弱酸性药物主要和白蛋白结合
- 弱碱性药物主要和a1-酸性糖蛋白结合
- 血浆蛋白结合影响效果
- 限制药物作用速度(只有游离的药物才能到达靶点发挥作用)
- 结合越多,活性成分越少,代谢越慢
- 药物相互作用
- 两种高血浆蛋白结合率的药物
- 相互竞争,结合率更高的药物挤占位点,使得结合率较低的药物释放,提高游离浓度引起药物中毒
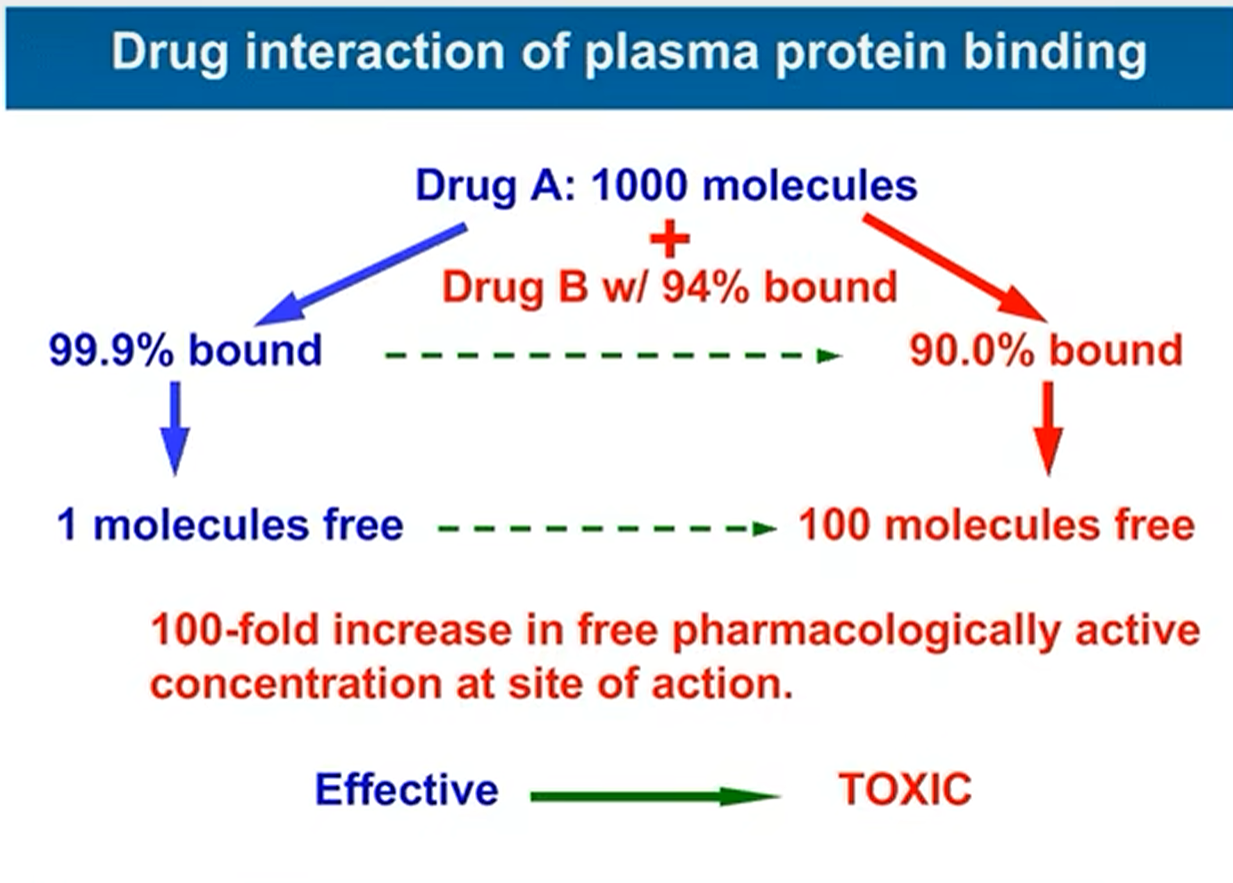
- 相互竞争,结合率更高的药物挤占位点,使得结合率较低的药物释放,提高游离浓度引起药物中毒
- 两种高血浆蛋白结合率的药物
- 血浆蛋白结合特点
-
血脑屏障(Blood brain barrier)
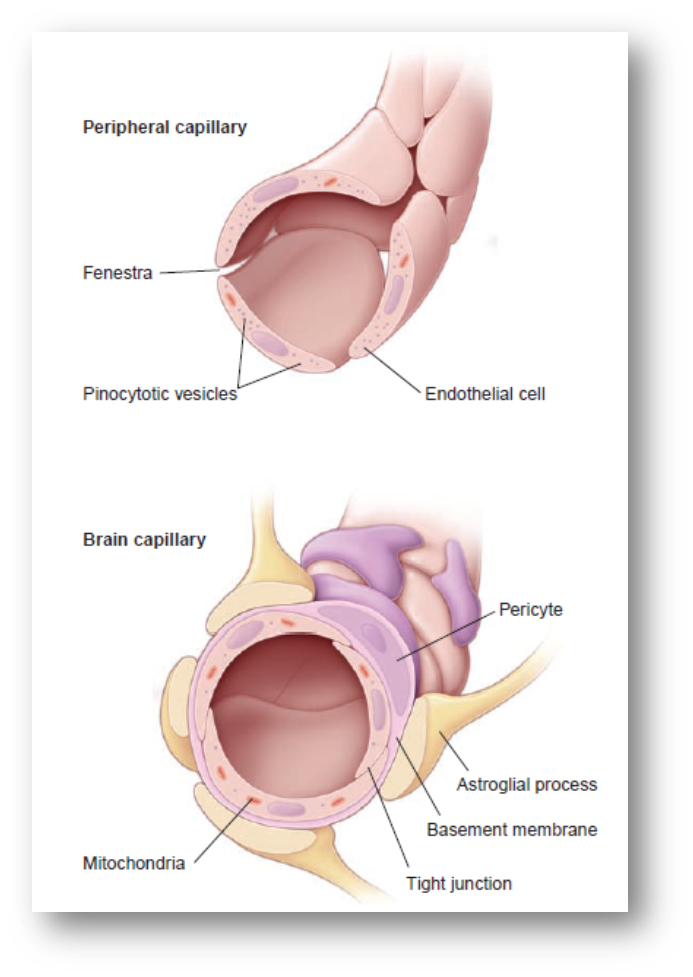
- 特点
- 大脑附近的毛细血管上皮细胞相互紧密连接形成的致密结构
- 仅有小分子强脂溶性的药物才能进入大脑
- 特点
大部分的药物都是弱酸或者弱碱
- 弱碱性药物在酸性环境下能离子化,更容易发挥作用,难以离开对应细胞区域
- 弱酸性药物在碱性环境下也能离子化,反之不能
- 胎盘屏障(Placental barrier)
- 和普通毛细血管上皮效果一样,对药物递送没有屏障效应
c. Metabolism, Biotransformation
-
代谢(Metabolism): Enzymatic alteration of drug molecule
-
代谢部位(site of metabolism)
- 大部分药物会被转运到肝脏进行降解
- 也可能发生在其他组织:肾脏、血浆和肠粘膜
-
代谢相(phases of metabolism)
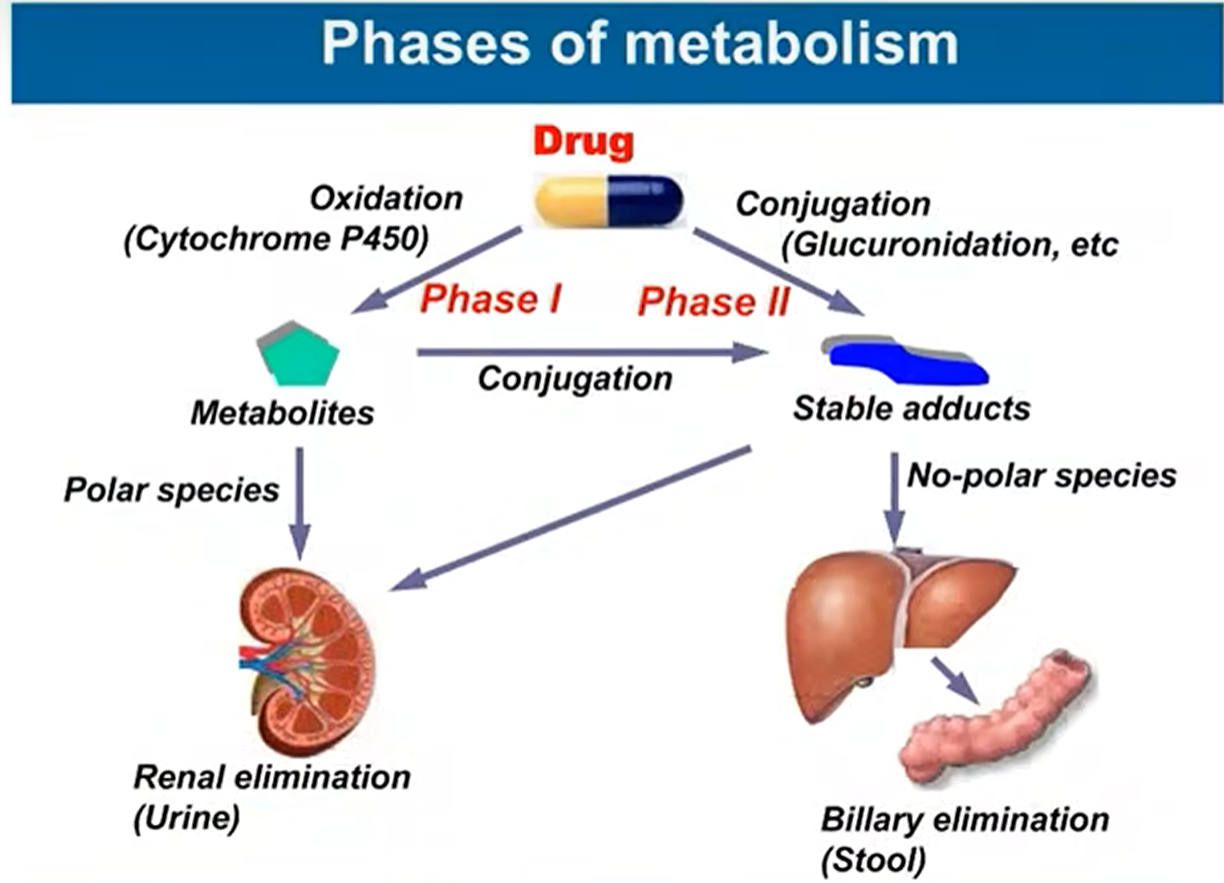
- 大部分药物都要经过氧化还原和二相结合变成水溶性强极性强的药物——极性强药物在经过肾脏之后,不容易被重吸收可排出体外;而脂溶性强了容易被重吸收难以代谢出体外
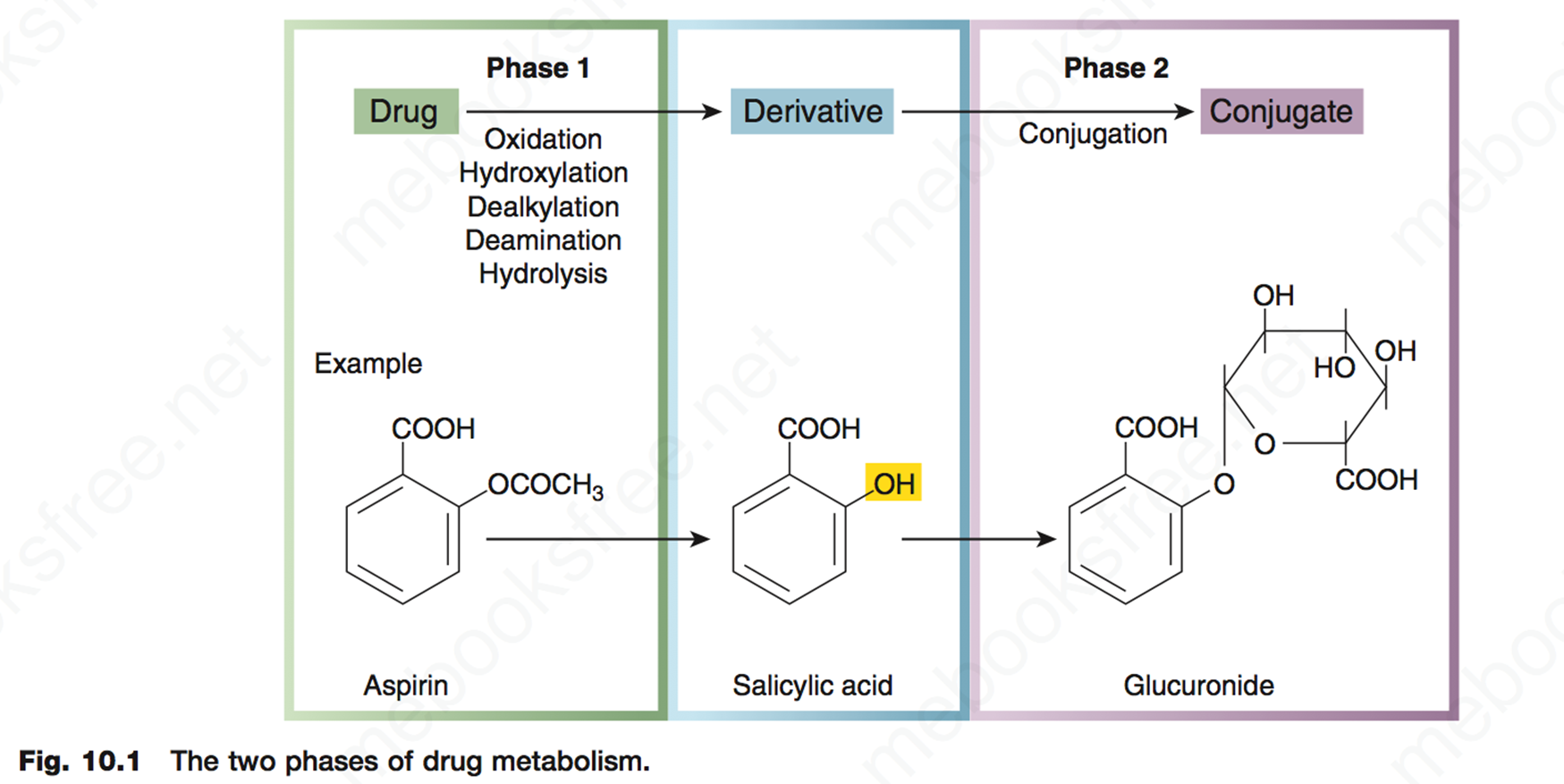
- 大部分药物都要经过氧化还原和二相结合变成水溶性强极性强的药物——极性强药物在经过肾脏之后,不容易被重吸收可排出体外;而脂溶性强了容易被重吸收难以代谢出体外
药物代谢的生物学意义:让药物排出体外,获得活性产物,灭活药物中止过强药效
-
肝脏对药物代谢的作用
- Phase I
- Product more reactive and introduce a functional group(-OH,-SH,-NH2)
- Phase II
- Addition of a large polar substituent(glucuronide sulfate or acetyl group)
- Meaning
- Decrease pharmacological activity
- Decrease lipid solubility -> increased rate of excretion
- increase the molecular weight
- Phase I
-
肝脏代谢引起的毒理
- 肝脏代谢并非总是走向解毒的代谢通路
- Phase I的代谢产物可能比母药更加具有生物毒性
- Phase II一般总是解毒步骤,但是也有可能性产生有毒物质
常见Phase I代谢的有毒产物一般都是亲电子基团
-
肝药酶(细胞色素P450):是一组结构和功能相关的超家族基因编码的同工酶,主要分布于肝脏微粒体中,故称为肝药酶;P450是在450nm紫外吸收峰————CYP3A4为主要药物代谢酶
详见:
-
药物-药物相互作用的两种形式:酶的诱导和抑制
- 酶的诱导(Enzyme Induction)
- 概念:使得酶的活性增高药物代谢加速,本质是酶的诱导剂使得酶的表达增多(分子数加多)
- 结果: 代谢活性下降,血药浓度降低,首过效应增强,生物利用度下降。
- 一般药物,代谢被诱导,活性降低毒性降低药效减弱
- 前药,代谢被诱导,活性提升药效增强
- 常见的酶诱导剂:利福平、苯巴比妥
- 酶的抑制(Enzyme Inhibition)
- 概念:使得酶的活性受到抑制,降低了药物的代谢速度
- 区分:诱导和抑制
- 酶的诱导和抑制分子上不是简单的逆过程:
- 酶的诱导是在基因水平调控使得酶的分子的表达量增加,从而提高活性和代谢速度,响应速度比较缓慢,机体相对容易调整
- 酶的抑制是在蛋白质分子水平直接抑制其活性位点降低酶的生物活性,响应速度非常迅速,机体不容易调整
- 酶的诱导和抑制分子上不是简单的逆过程:
- 效果:母药(parent drug)浓度增加,减少代谢产物浓度,增强或延长药效,可能引起毒性————前药有效
- 酶的诱导(Enzyme Induction)
-
前药和软药
- 前药:也称前体药物、药物前体、前驱药物等,是指药物经过化学结构修饰后得到的在体外无活性或活性较小、在体内经酶或非酶的 转化释放出活性药物 而发挥药效的化合物
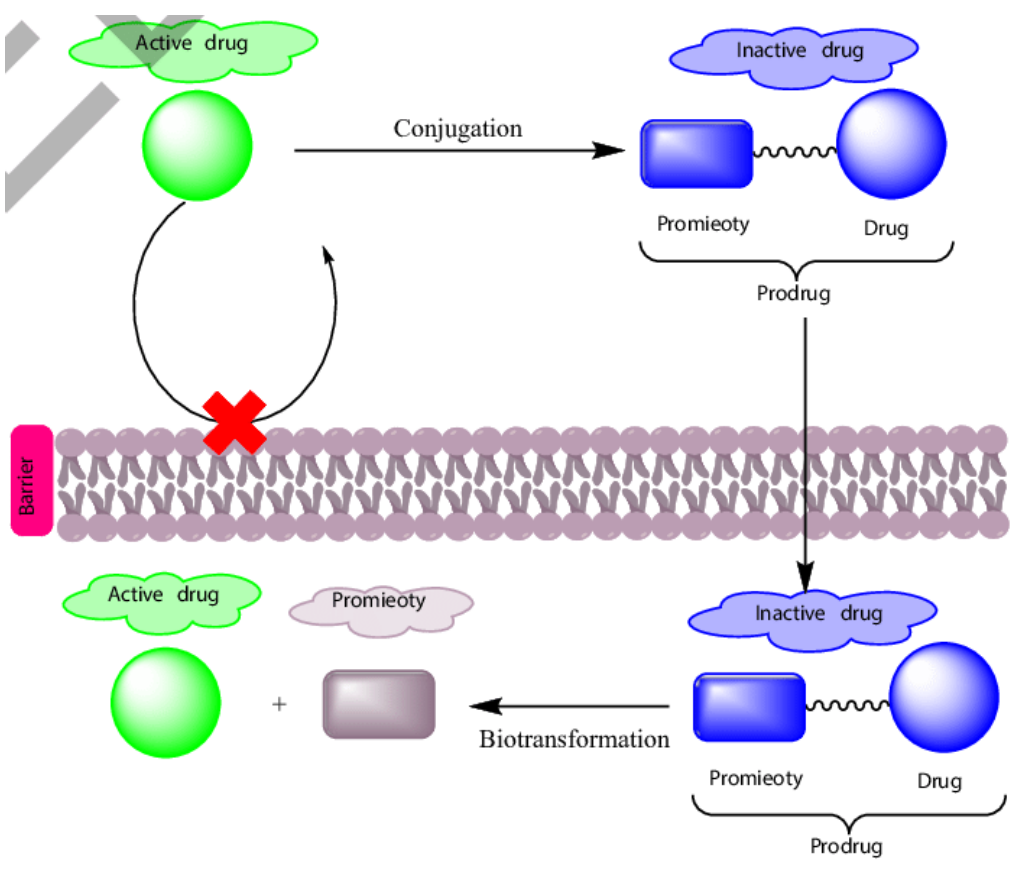
举例分析
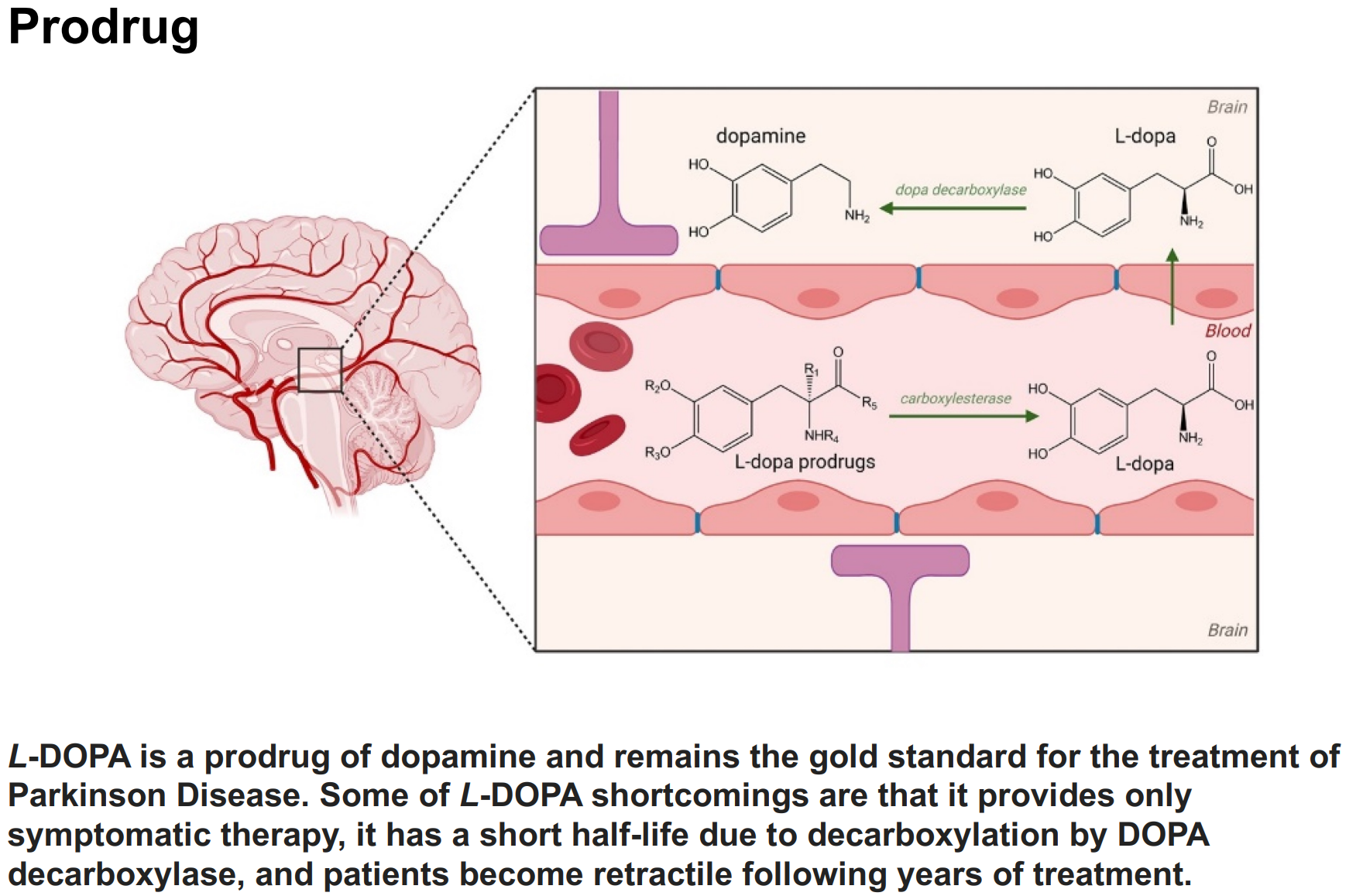
- 软药:是指药物本身母药结构具有药理活性,在发挥药效后 按照既定的代谢途径 迅速地被转化为不具有药理活性或毒性的代谢产物,从而实现在发挥药效的同时,避免原型药物、活性代谢物、反应性代谢物对机体造成毒性。
- 前药:也称前体药物、药物前体、前驱药物等,是指药物经过化学结构修饰后得到的在体外无活性或活性较小、在体内经酶或非酶的 转化释放出活性药物 而发挥药效的化合物
d. Excretion
-
排泄(Excretion): the process by which a drug or metabolite is eliminated from the body
-
排泄途径
- 肾脏(主要器官)
- 主要功能
1. 维持体液的水盐平衡稳态
2. 滤除代谢产物 - 肾单元的滤过系统
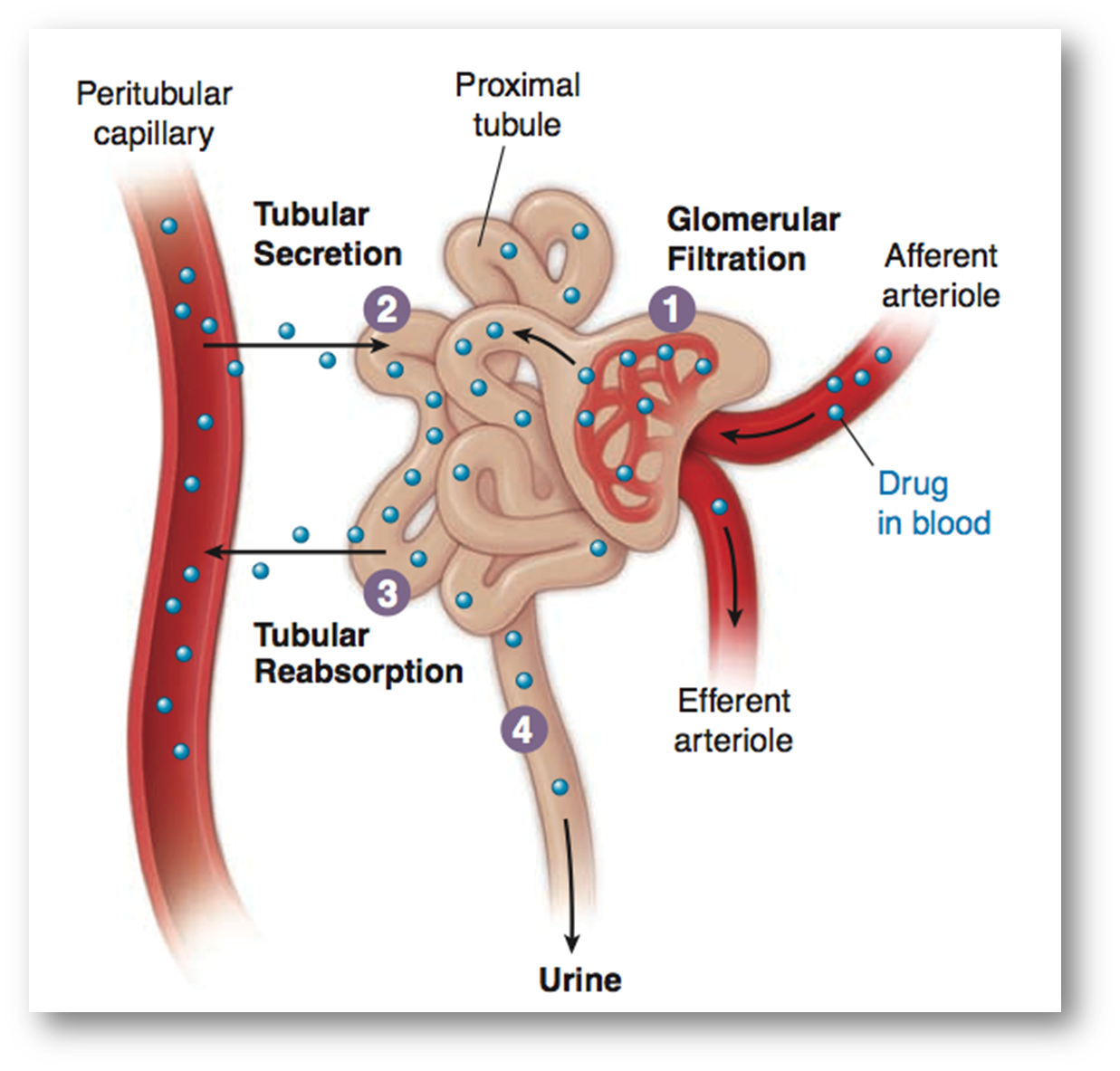
> 因此为了使得药物分子可以经过肾脏代谢,一般都需要该药物具有一定的亲水性 - 排泄步骤:滤过 -> 转运载体(分为阴离子通道————酸性药物和阳离子通道————碱性药物)-> 重吸收(调整ph来降低,增加解离型药物分子)
- 主要功能
- 胆道
- 肠肝循环:药物从肠道进入肝脏,代谢后通过胆道排泄进入肠道,在肠道重吸收进入血液
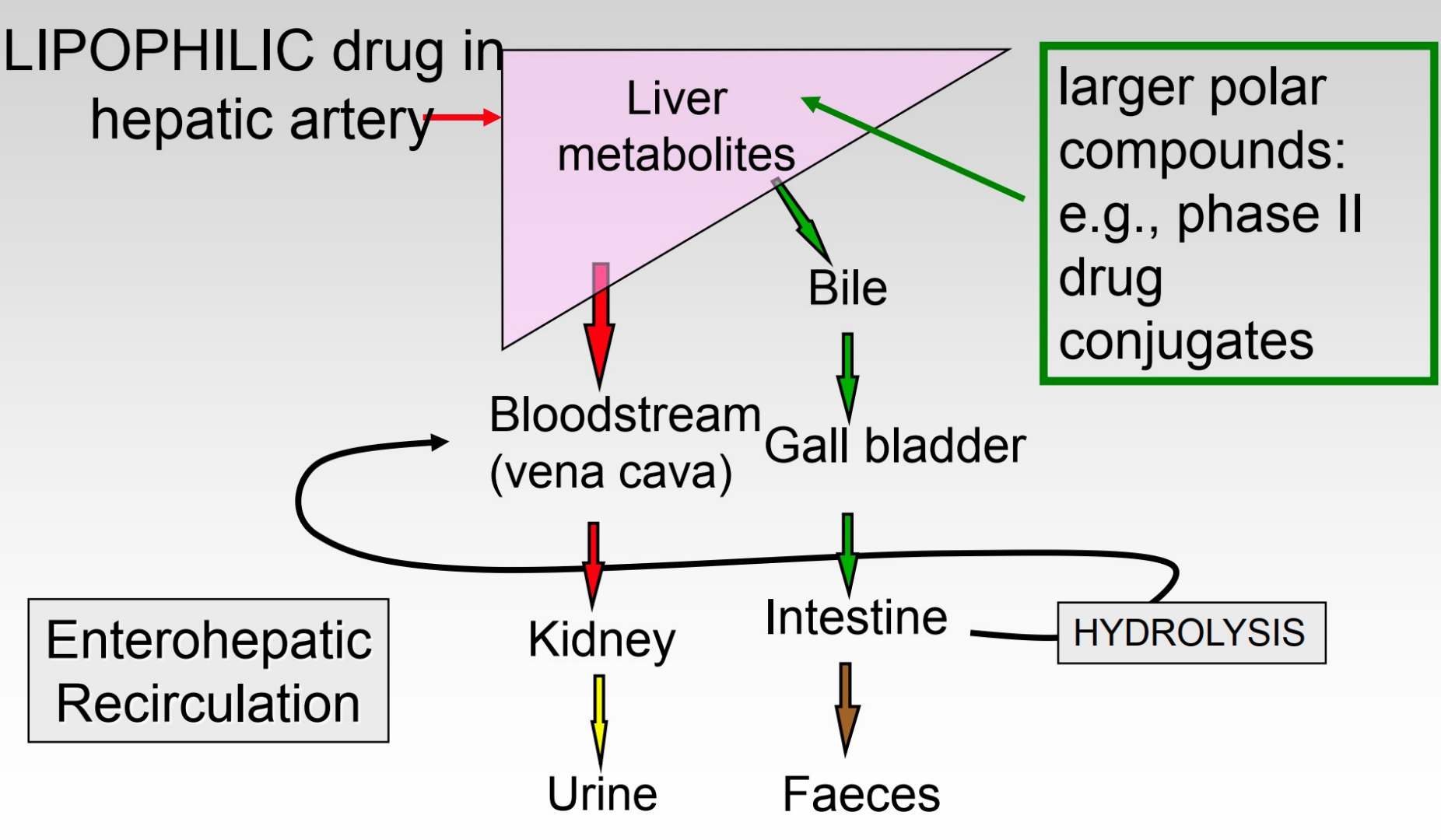
- 效果:延长药物作用时间
- 肠肝循环:药物从肠道进入肝脏,代谢后通过胆道排泄进入肠道,在肠道重吸收进入血液
- 呼吸、汗腺、唾液、腺泪腺、乳腺
- 肾脏(主要器官)
药物浓度时间过程 Time course of drug concentration
研究药物动力学主要目的:设计给药方案,达到有效浓度且不至于产生不良反应
药代动力学曲线:
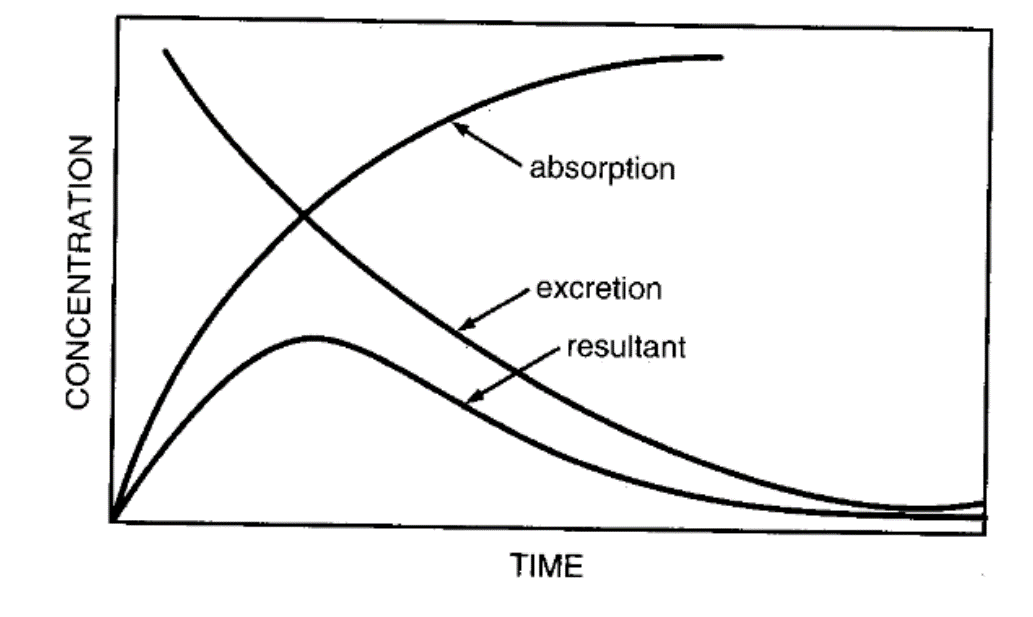
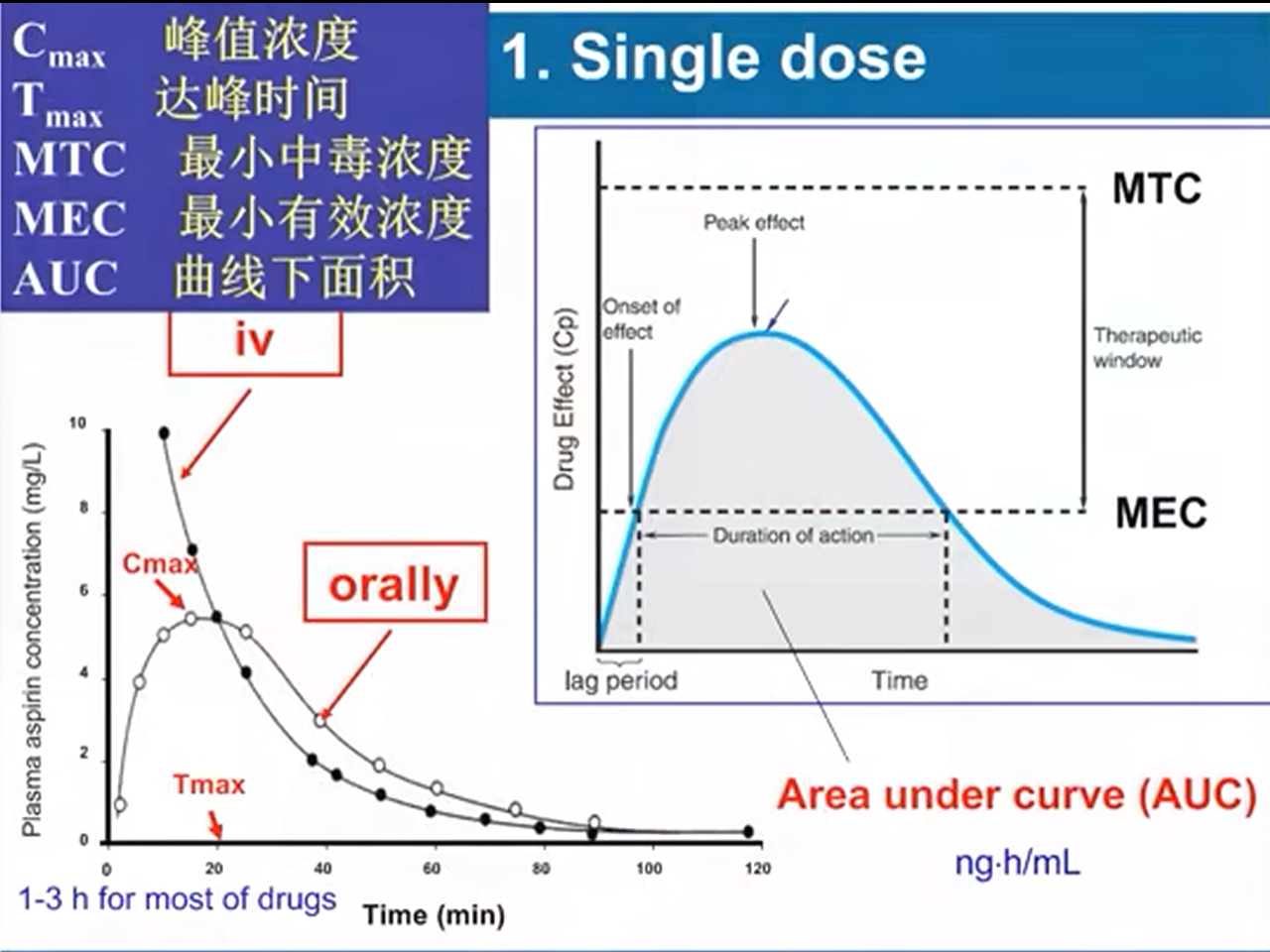
单剂量给药 single dose
- 血管内给药————持续降低的曲线
- 血管外给药————峰形曲线
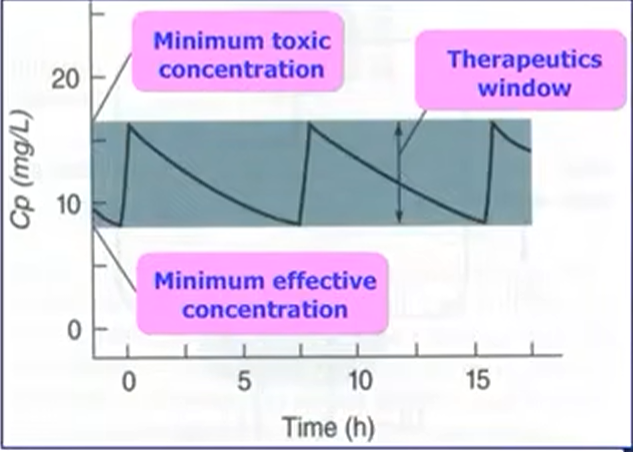
- MEC -> Minimum Effect Concentration
- MTC -> Minimum Toxicity effect
主要反映机体药效学指标
- Therapy Window: MEC< drug concentration < MTC
- Lag period: the minimum time to reach MEC
- T max: 达峰时间(反映药物吸收时间)
- C max:峰值浓度(速效药物峰浓度高;缓释药物峰浓度低)
- AUC:曲线下面积(反映药物吸收程度,在峰浓度附近效应最大可以监测生理指标)
- Bioavailability 生物利用度
- 概念:生物利用度(bioavailability ,BA),描述药物被吸收进入人体循环的速度与程度。
- 计算公式:

- 进入循环系统(血浆)的药物浓度总量可以计算曲线的AUC
静脉注射生物利用度一定是1,口服等其他方式小于1
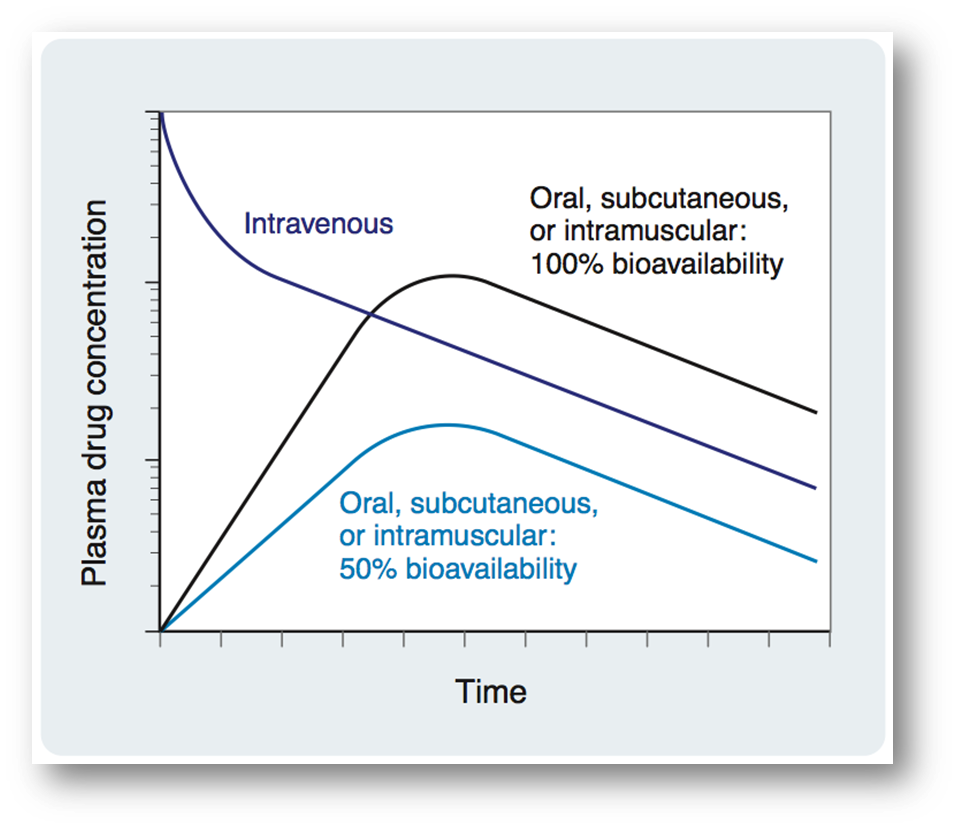

- 影响因素
- Destroyed in gut
- Not absorbed
- Destroyed by gut wall
- Destroyed by liver
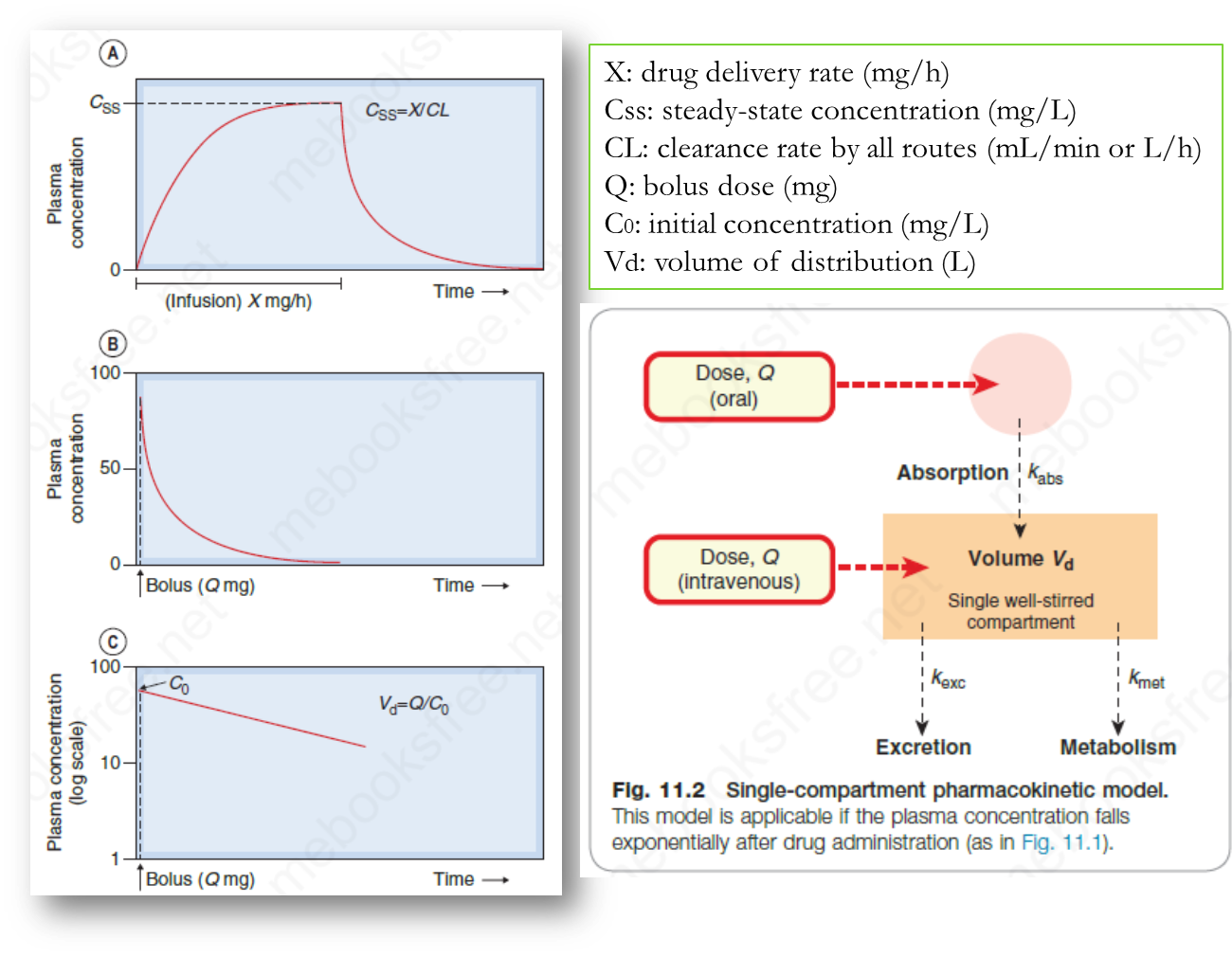
- Apparent Volume Distribution 表观分布容积
- 概念:剂量和浓度的比值,反映了药物在溶剂中的分散程度
- 计算公式:
- 表观容积反映药物的分布特性,值越大,药物扩散能力越强,药物在血浆中的分布浓度越低,药效越广;值越小,药物扩散能力越弱,药物在血液中的分布浓度越高,药效越受限
- 举例:
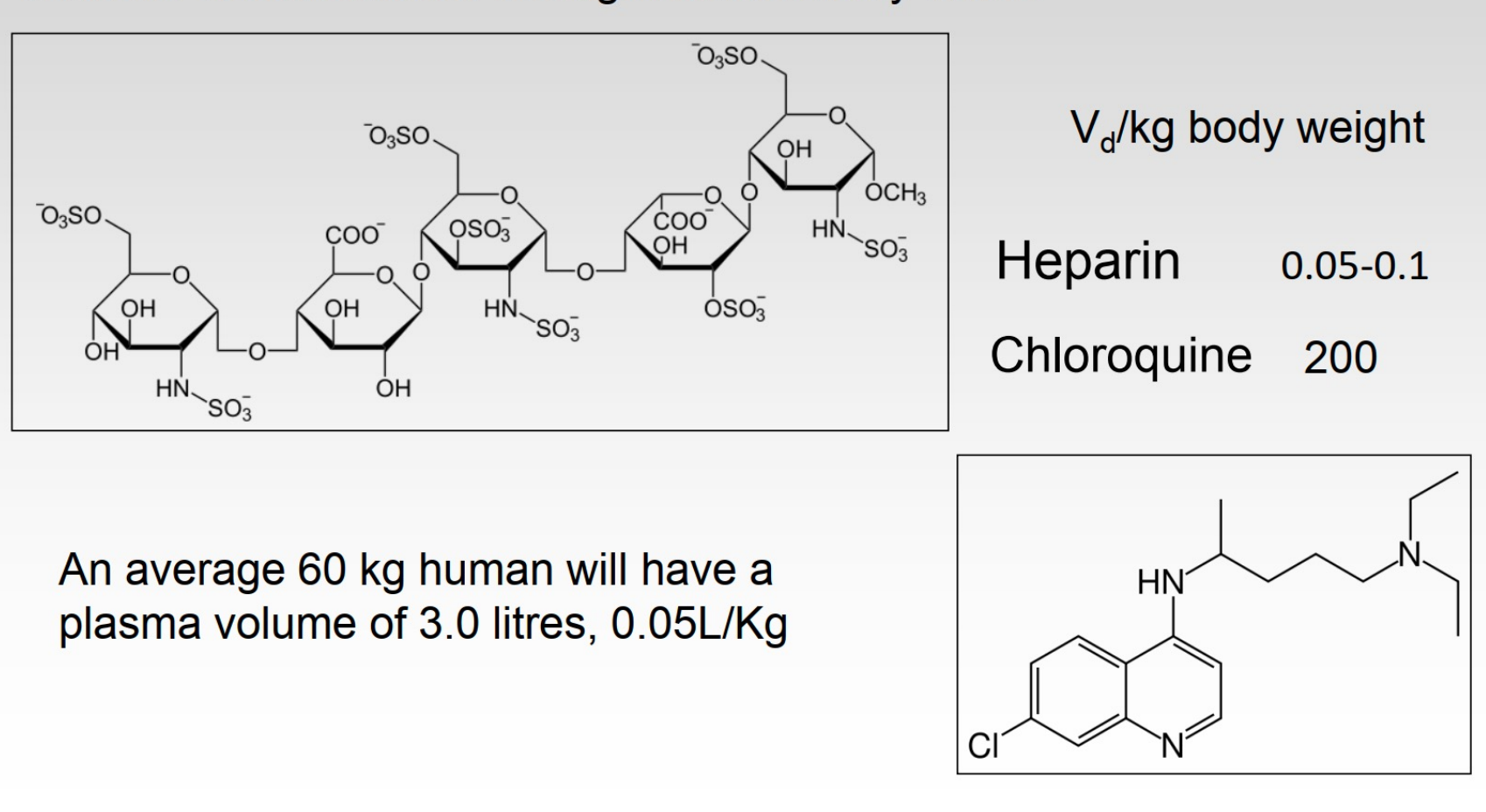
- 举例:
- 表观分布容积的最小值是血容量
- 表观分布容积的应用
- 评估药物的分布情况
- 计算给药剂量
- 房室模型

判断是一室还是二室模型通过浓度时间曲线来决定
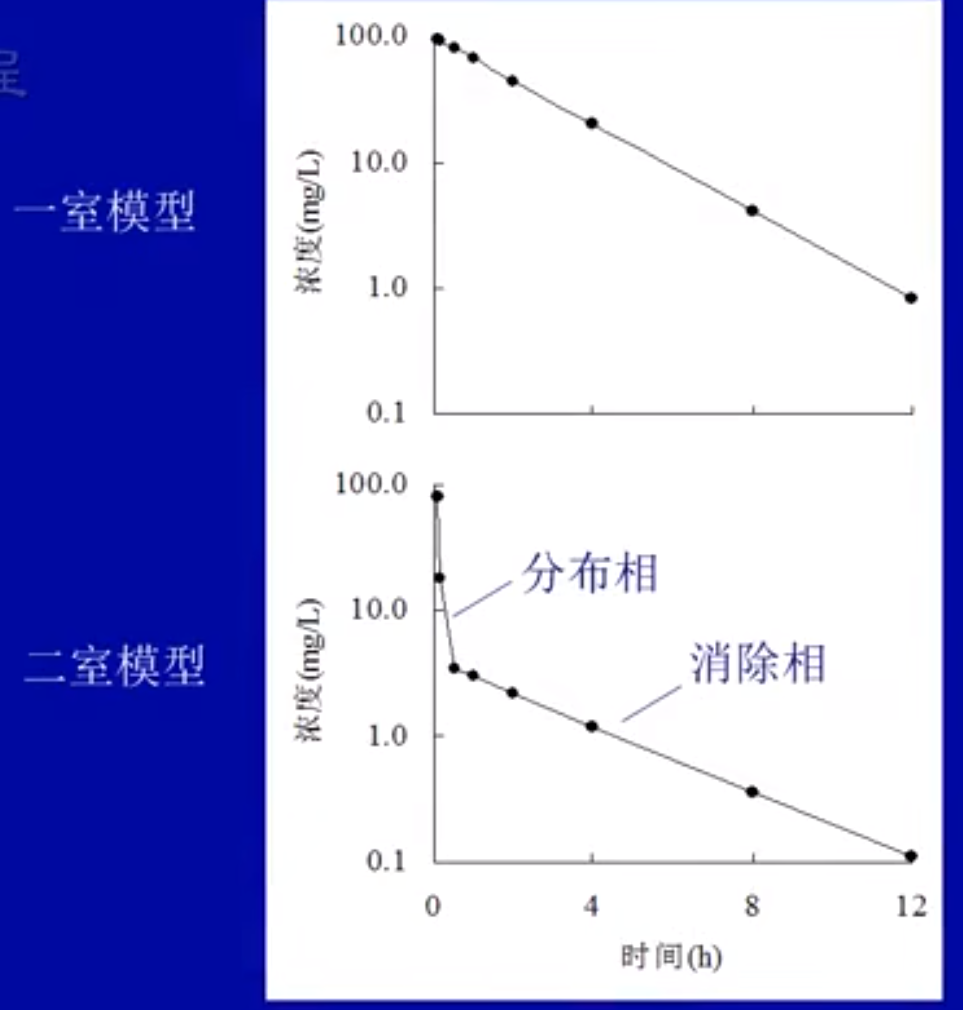
- Single-compartment model 一室模型
- Single-compartment model 一室模型
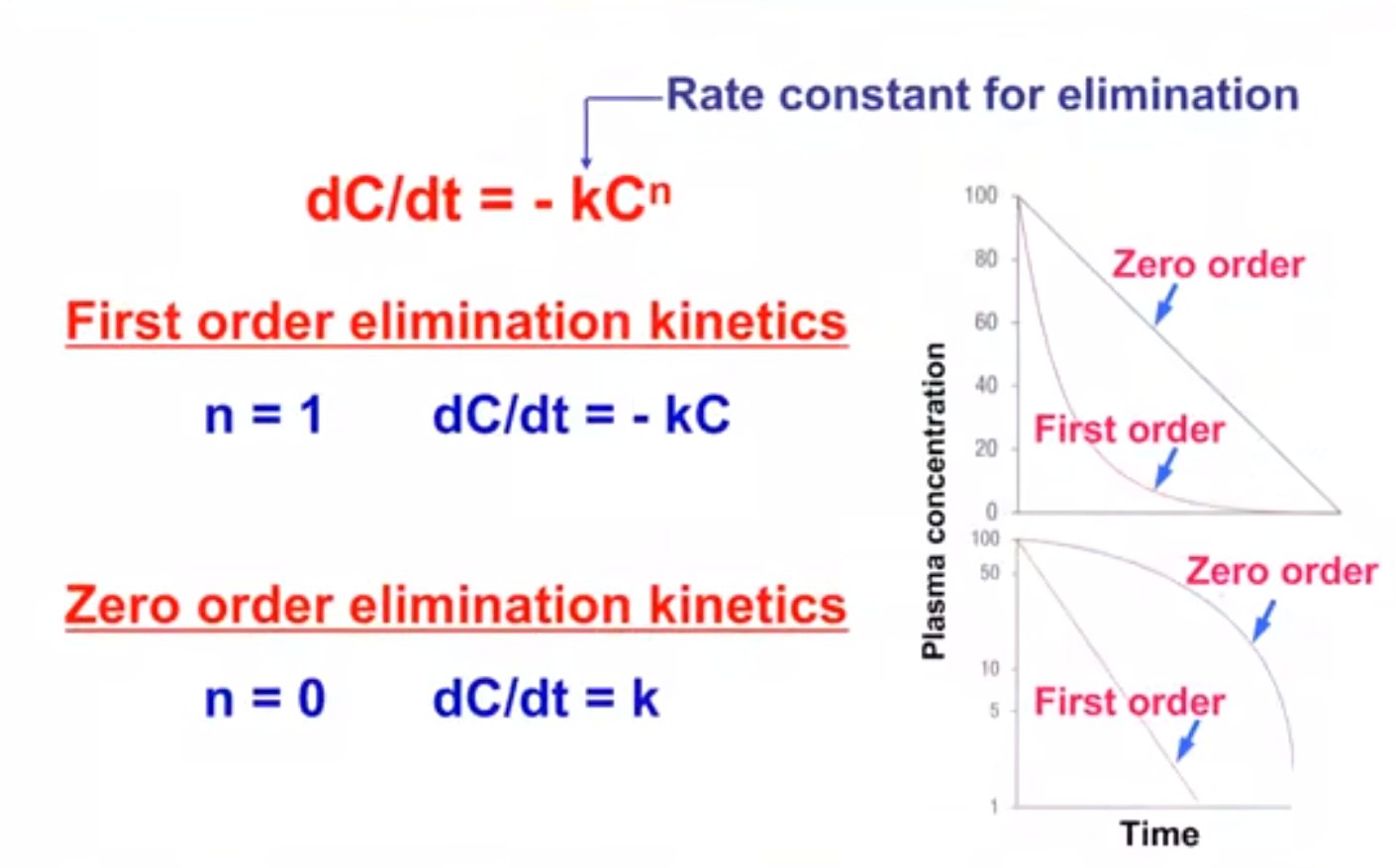
Elimination kinetics 消除动力学
-
速率过程
-
一级动力学(恒比消除)
- 药物在单位时间内以 恒定比例 转运或者消除
- 单位时间内消除速度和当时血药浓度成正比
- 大部分药物在治疗剂量下符合一级动力学过程
- 原因:大多数药物跨膜转运规律是脂溶扩散,符合菲克定律——由浓度梯度决定
-
零级动力学(恒量消除)
- 药物在单位时间内以 恒定药量 转运或者消除
- 任何时间的消除速度与当时血药浓度无关
- 有饱和机制(酶、载体),存在位点最大量;最大限度以内为一级动力学
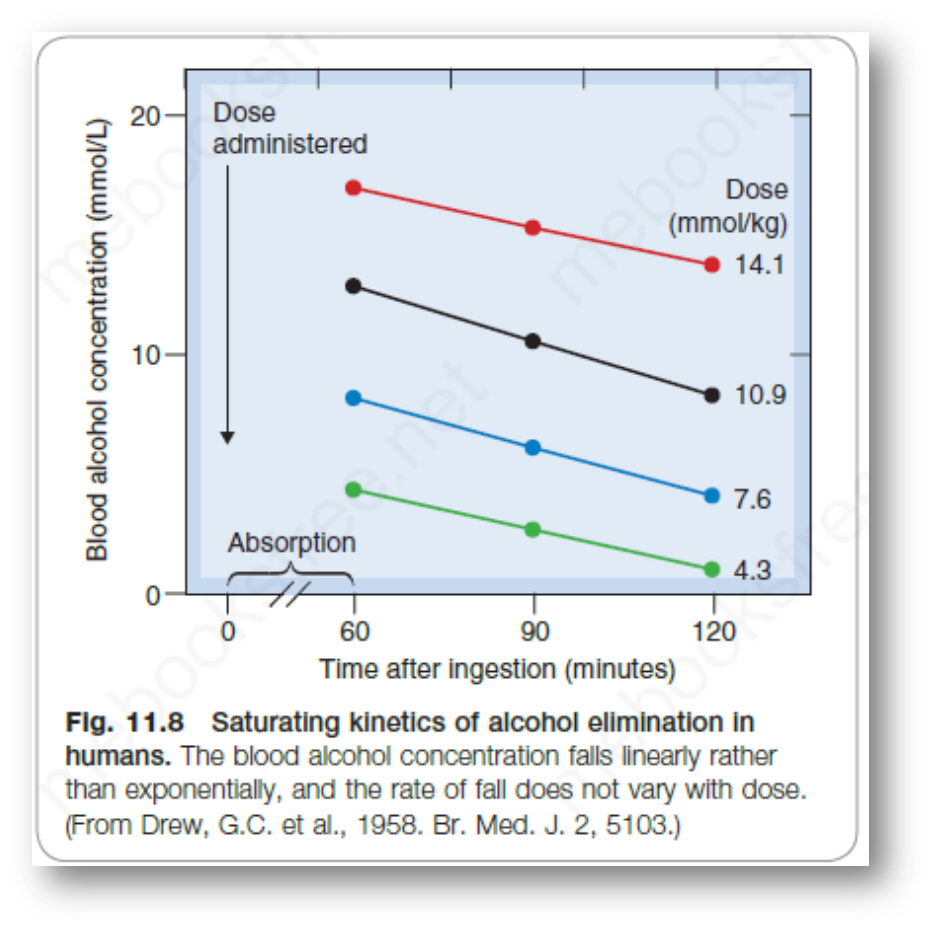
-
非线性速率(米氏动力学)
- 药物在高浓度时候为零级速率
- 药物在低浓度时候为一级速率
现代药物由于药物活性较高,一般使用剂量非常微量,一般都满足一级速率
-
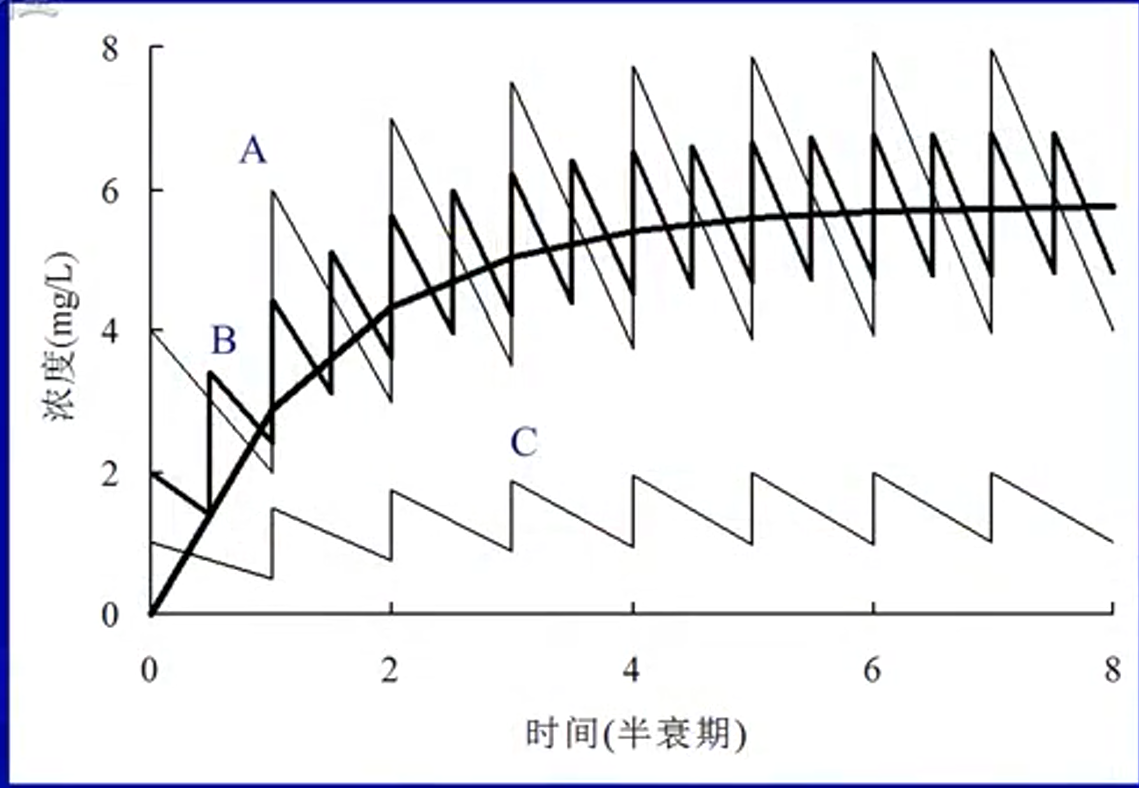
治疗剂量和频率对药物浓度的影响
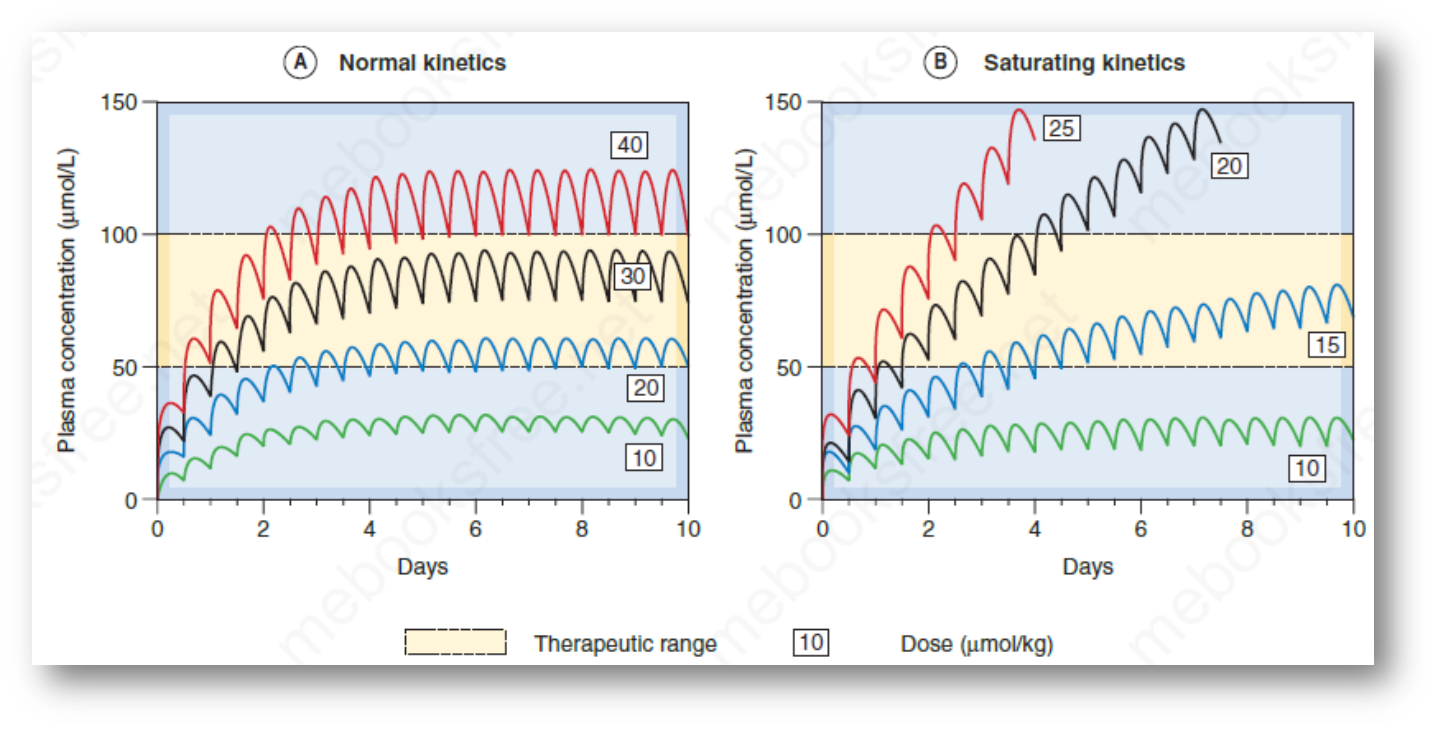
- A图:在单次低剂量下,随着持续给药,药物浓度能够逐渐趋近于达到稳态浓度
- B图:在单次高剂量下,由于代谢酶达到饱和,随着持续给药,药物浓度呈线性增长
剂量的高低,视特定药物在身体中的代谢能力而定
-
-
半衰期(t1/2)
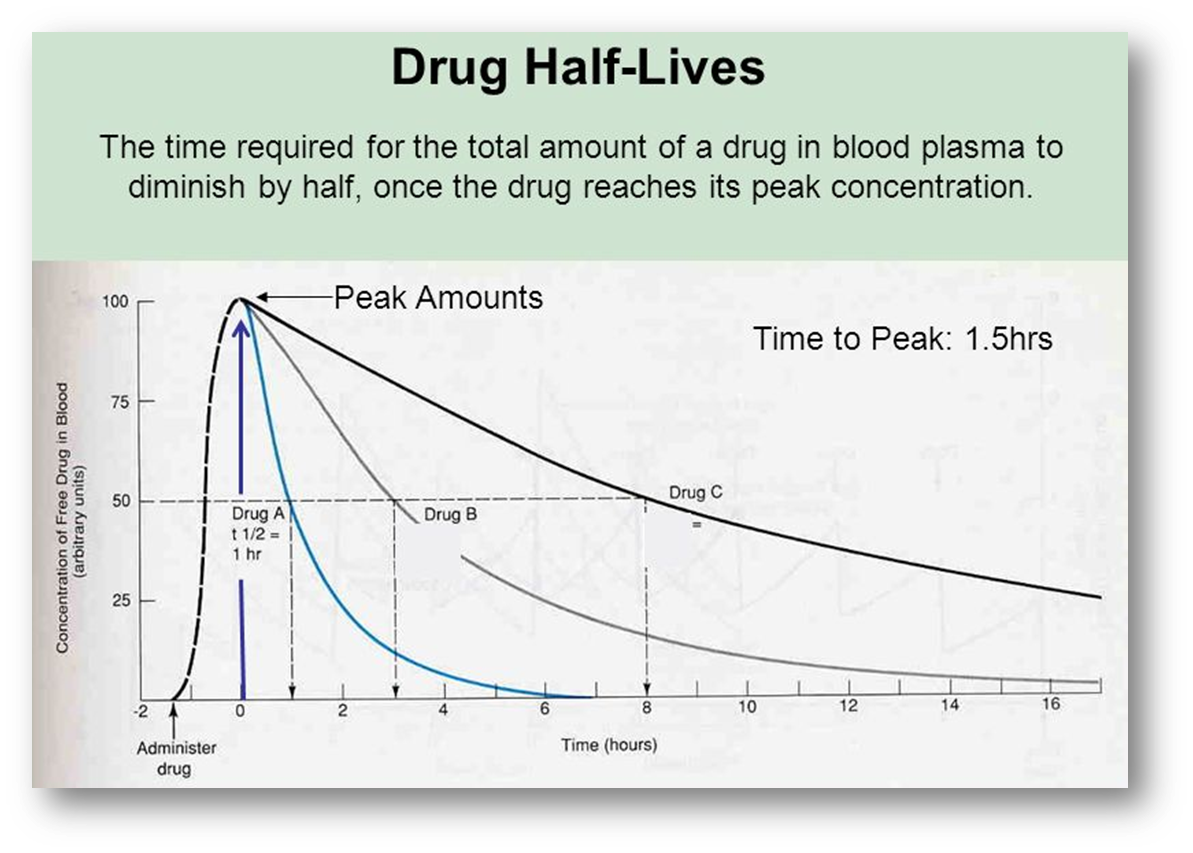
- 血药浓度下降一半所需要的时间
- 反映药物的消除程度
- 确定给药间隔的依据
- 测算达到稳态的时间和药物清除时间
-
清除率(CL)
- 单位时间内能把多少容积血中的药物全部清除
- 计算:消除速度/血药浓度
多剂量给药 Multiple dose
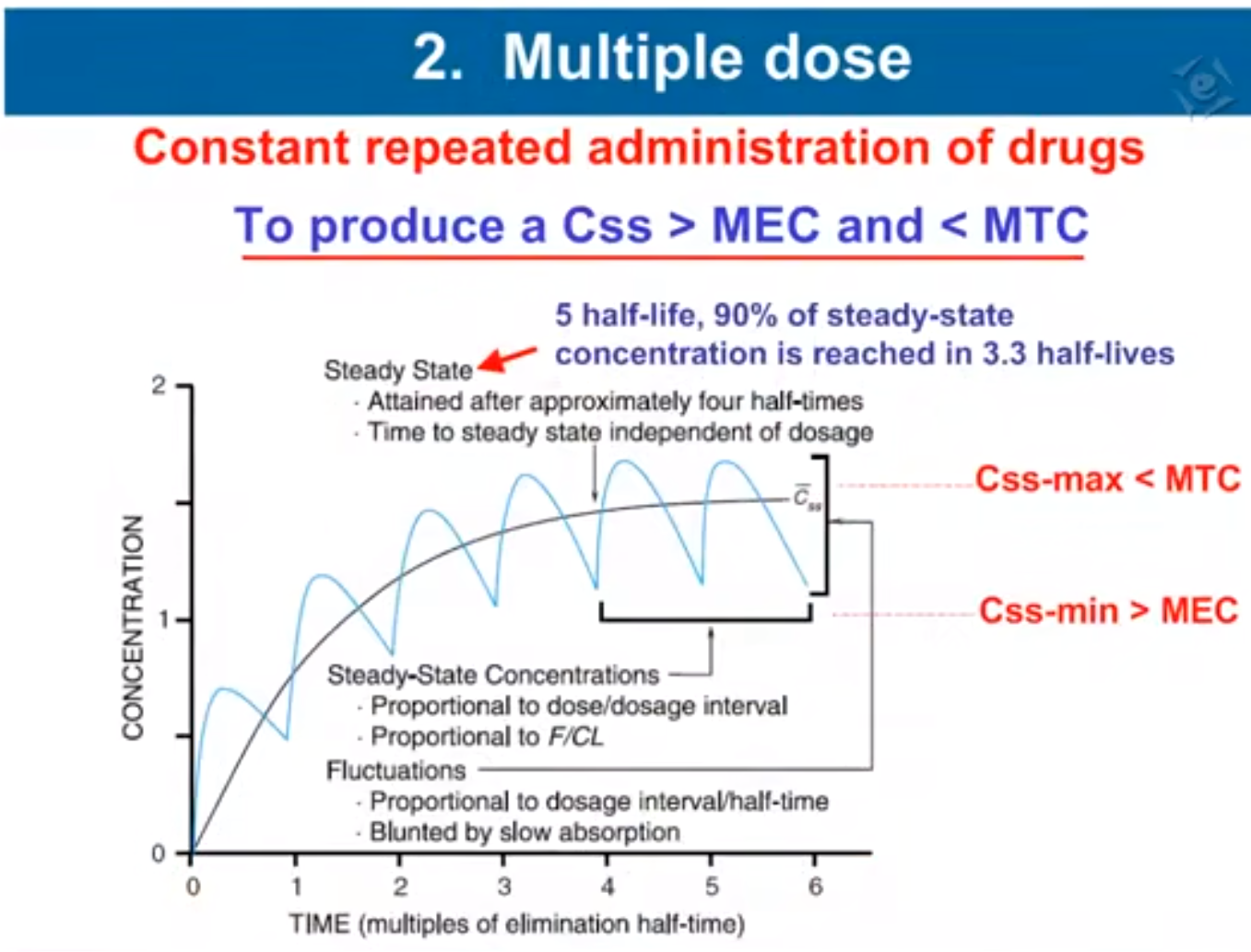
- 治疗窗口窄的药物为了维持稳定血药浓度以达到治疗效果同时不会中毒,一般采用静脉点滴的方法来精准控制
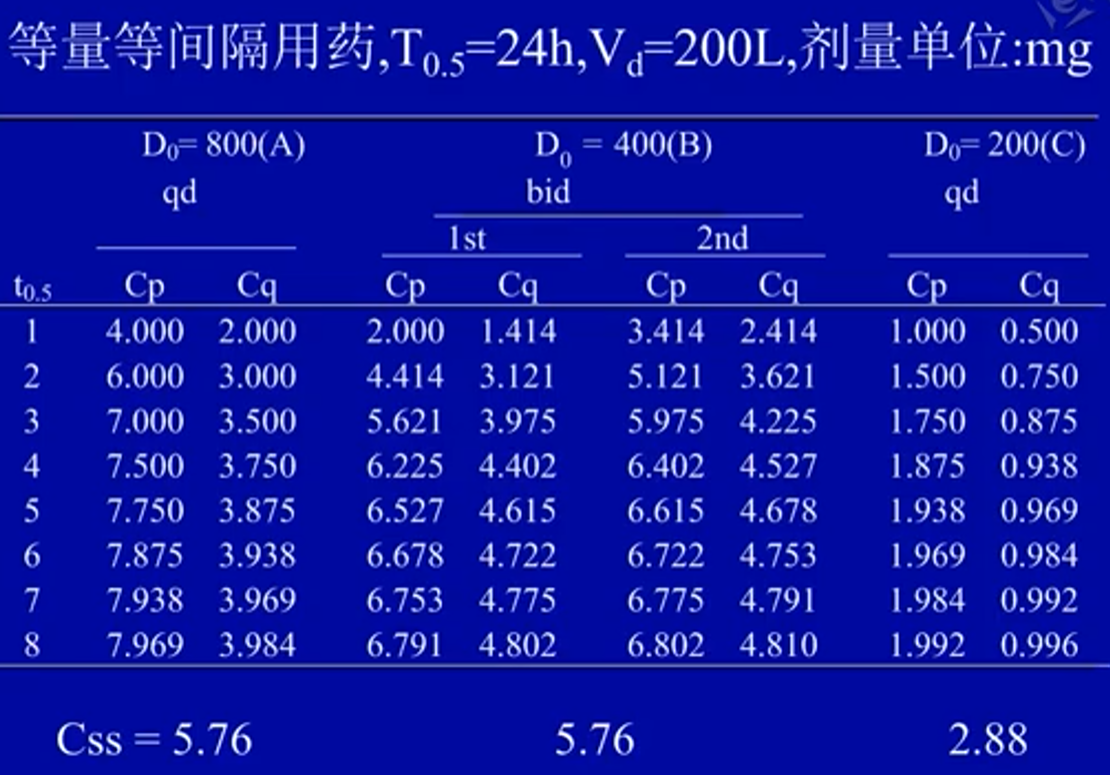
- 给药次数与血药浓度的关系
- 相同给药浓度,给药频率越少浓度波动越大
- 相同给药频率,给药浓度越低达到稳态的时的药物浓度越低
多次用药稳态(steady-state)
- 等量等间隔给药时,血药浓度逐渐上升,经过5个半衰期后达到稳态
- 进出平衡:在一个给药间隔内清除药量等于进入体内药量
- 水平波动:血药浓度在平均浓度上下波动
坪值(plateau)浓度 != 峰谷平均 = 等于峰形曲线面积/时间
- 等量等间隔多次给药的血药浓度变化规律
- 平均稳态浓度高低 正比于 每个半衰期给药总量
- 血药浓度波动大小 正比于 每次药量(总量不变)
- 达坪时间只与药物半衰期有关,恒为5个半衰期
缓释制剂的原理
相比于普通制剂,类似于静脉推注,药物迅速崩解被吸收;缓释制剂相当于静脉点滴,逐渐缓慢释放药物
仍存在微小的药物浓度波动,可使得血药浓度相对平稳
相比于普通同种药物少量多次,缓释制剂还是从给药方便角度设计。其最终效果是等效的
负荷量/维持量给药 Loading dose
- 首次剂量加大,然后给予维持剂量(Maintenance),使稳态治疗浓度提前产生
- 通常在需要立即使血药浓度达到稳态浓度而快速起效时采用
如磺胺药物按照半衰期间隔给药首次剂量加倍,原因是便于快速达到稳态浓度
如果治疗窗口小的药物也可以采用分次加倍给药————在一个半衰期内固定给药剂量,分多次加倍剂量,使得总剂量是之前两倍,单次血药浓度不会太高而造成中毒
- 为什么氯吡格雷的负荷量(300mg)是维持量(75mg)的4倍?
重要的PK参数
- 吸收: AUC(吸收程度)、C max(达峰浓度)、T max(吸收速度)、BA(生物利用度)
- 分布: Vd(表观分布容积),房室数(反映分布速率)
- 消除: 半衰期、清除率
药效学(Pharmacodynamics)
- 药物如何对身体起作用
基本概念(引自Cytc的APH202笔记)
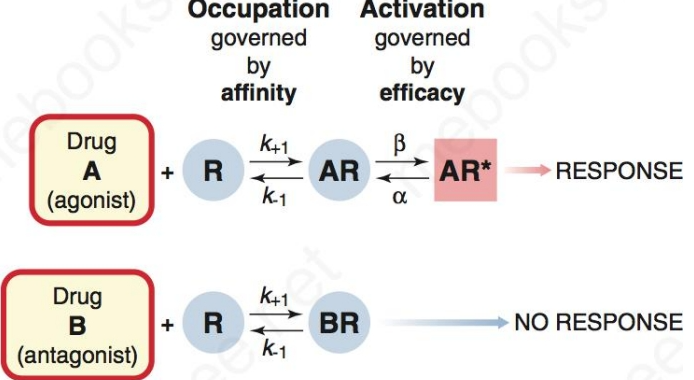
激动药(agonist)
-
亲和力(Affinity)与效能(Efficacy)
- 亲和力所描绘的是药物与受体的结合趋势,这一特性与其效价强度(potency,引起等效反应的药物浓度或剂量)成正比
- 效能所描绘的则是药物对受体的激动能力,用来表述
- 亲和力能评估拮抗药和激动药的特性,但是效能只适用于激动药
亲和力也可以用结合系数描述
Inhibitory Constants
-
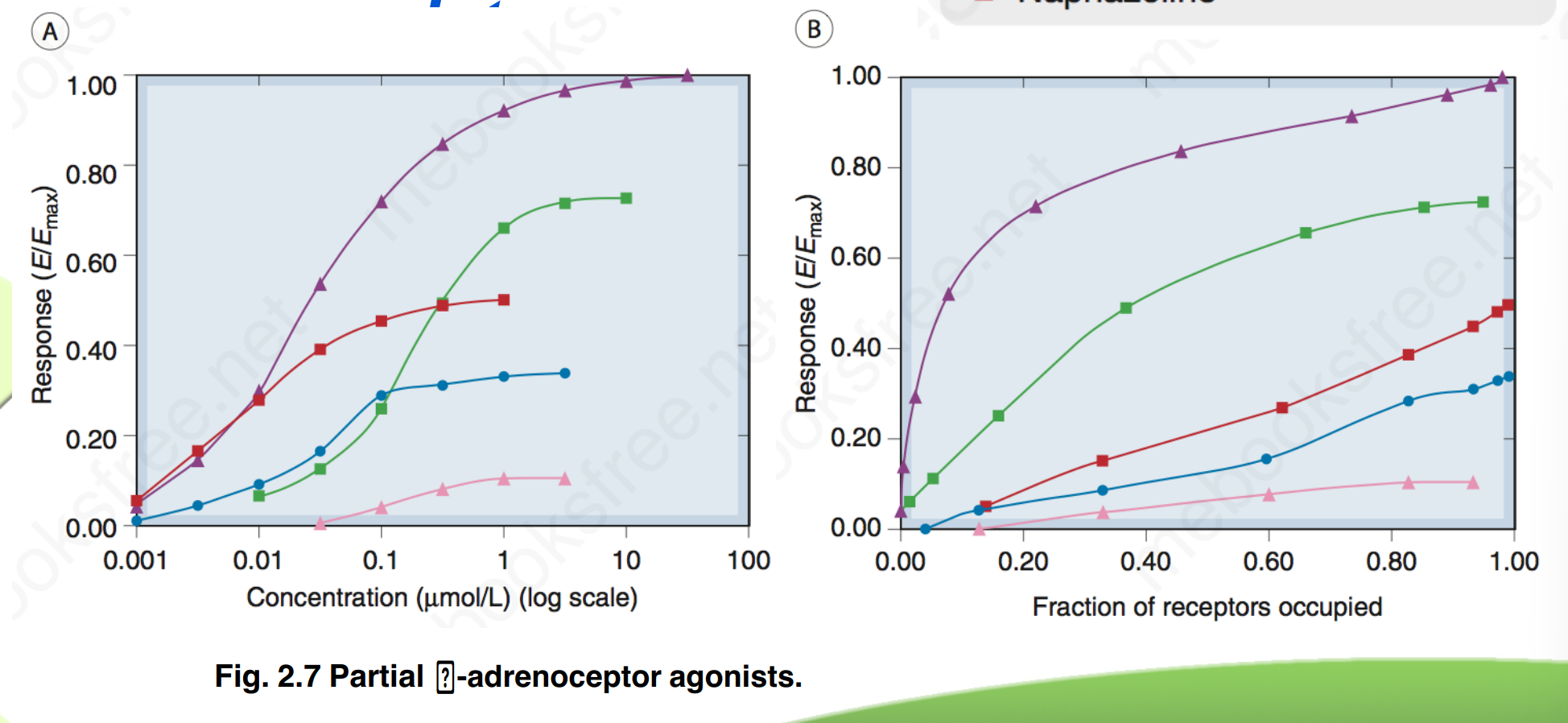
完全激动药(Full agonists)、部分激动药(Partial agonists)和反向激动药(Inverse agonist)
- 完全激动药β=1,可以在占领全部受体后产生最大化效应,具有高效能
- 部分激动药β<1 ,永远无法达到最大化效应,效能较低,与完全激动药合用时还会拮抗部分效应
- 反向激动药使被激活的受体失活的药物,即所产生效能为负
-
别构药物(Allosteric drugs)
- 所结合位点与内源化合物与受体的结合位点不同的药物,通过引起受体结构变化,同时影响激动药的亲和力与效能,可正向可负向
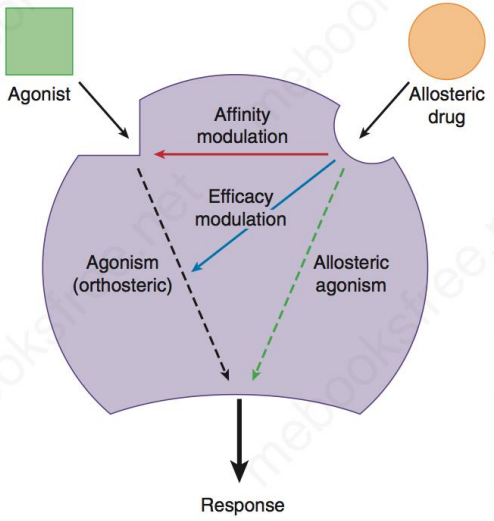
- 所结合位点与内源化合物与受体的结合位点不同的药物,通过引起受体结构变化,同时影响激动药的亲和力与效能,可正向可负向
拮抗药(antagonist)
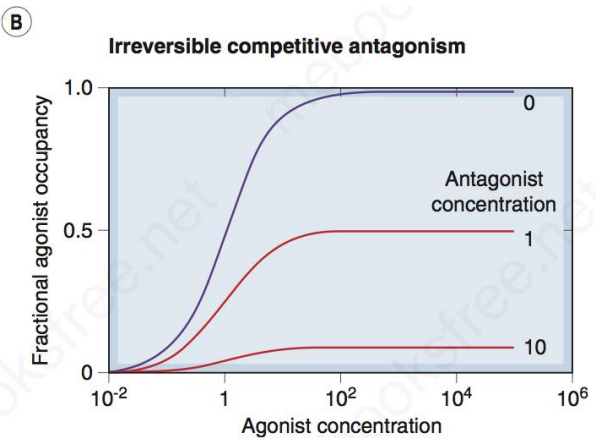
- 竞争性拮抗(competitive antagonism)
- 产生此类药效的拮抗药与激动药竞争相同受体位点
- 可逆拮抗药(非共价结合):增加剂量竞争的激动药最大效能不变,但是亲和力(效价强度)降低
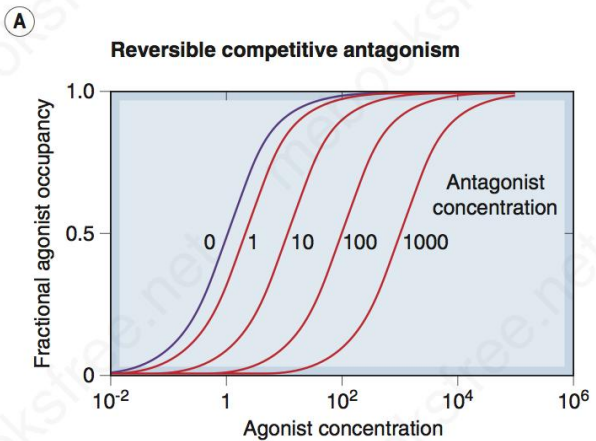
- 不可逆拮抗药(共价结合):增加剂量竞争的激动药最大效能和亲和力均降低
- 非竞争性拮抗(non-competitive antagonism)
- 产生此类药效的拮抗药与激动药竞争相同不
同受体位点,为别构结合。对于其所竞争的激动药产生的效果类似于不可逆的竞争性拮抗
药
- 产生此类药效的拮抗药与激动药竞争相同不
受体理论 & 药物效应动力学
药效学基本概念
- 药物研究阶段 (according to drugability)
- 确定药靶 <- 候选药靶 <- 潜在药靶 <- 功能蛋白
- 主要常见靶点:受体、酶、转运体和离子通道
- 药物的基本作用
- 不产生新功能
- 药物通过调节机体组织器官原有生理生化功能发挥作用
- 药物基本作用:兴奋、抑制
- 区分药物作用和效应(Action & Effect)
- action = how drug works(初始作用)
- effect = consequence of drug action
- 区分局部作用和吸收作用
- 局部作用:药物没有吸收入血,就在给药局部发挥作用
- 吸收作用:进入血液循环对全身各大系统产生的影响
- 有药物希望局部给药产生局部作用,所以剂量很低能快速被代谢,来实现仅在给药局部发挥作用;有的药物希望局部给药产生吸收作用,采取注射方法,希望延长作用时间。
- 药物作用的四个水平

在不同作用水平上可能表现处不同的兴奋与抑制,举例分析:
- 吗啡在功能层面上是镇痛,抑制神经系统的功能。但在分子层面是阿片受体的激动剂,提高了内源性镇痛系统的功能增强
- 阿司匹林(非甾体类抗炎药物)在分子层面抑制前列腺素合成酶。在功能层面达到解热镇痛的效果
前列腺素:引起炎症疼痛发烧反应的主要体内活性物质
- 药物作用的两重性
- 治疗作用(therapeutic action)
- 对因治疗(Etiological treatment)
- 对症治疗(Symptomatic treatment)
- 补充治疗(Supplementary treatment)
> 由于医药发展比较缓慢,对于大部分疾病的病因了解不够深入,或者没有针对的治疗手段,所以现有超过70%的药物都还是对症治疗
> 如:抗感染、抗寄生虫药物主要是对因治疗;高血压糖尿病相关药物主要是对症治疗,能长期控制,但是难以治愈
- 不良反应(Adverse drug reaction, ADR)
- 与用药目的无关,并造成对病人不利的反应
- 副作用(side effect)
- 在治疗剂量时,出现与治疗目的无关的不适反应(会随目的变化而变化)
- 举例: 阿托品用于散瞳,也会抑制腺体分泌
- 在治疗剂量时,出现与治疗目的无关的不适反应(会随目的变化而变化)
- 毒性反应(toxic reaction)
- 剂量过大或蓄积过多发生的危害性反应(有个体差异,高敏感低剂量也会有反应)
- 致畸、致癌、致突变
- 剂量过大或蓄积过多发生的危害性反应(有个体差异,高敏感低剂量也会有反应)
- 后遗效应(residual effect)
- 停药后需要浓度已降至阈值一下依然残存的生物效应
- 停药反应(withdrawal reaction)
- 停药后原有疾病加重
- 变态反应(过敏反应,allergic reaction)
- 机体受药物刺激后发生异常的免疫反应(与原有效应,剂量无关)
- 特异性反应(idiosyncrasy)
- 与遗传有关,反应性质可能与常人不同(不可预期,与药理作用无关)
- 继发性反应:由于药物治疗效果引起的不良后果
- 如:抗菌药物抑制细菌造成白色念珠菌(真菌)大量繁殖造成的二重感染
- 治疗作用(therapeutic action)
靶点(target)
- 概念:体内能够与特定药物特异性结合并产生治疗疾病或调节生理功能作用的生物大分子
药物受体相互作用 Drug-Receptor Interaction
-
受体 receptor
- 特性:大分子,特异性结合产生效应
- 接受第一信使,并通过直接或间接产生第二信使传递信号
-
配体 ligand
- 和受体特异性结合的物质
-
药物-受体相互作用的特征
- 形成化学键
- 存在饱和性、竞争性
- 具有特异性
- 构效关系:受体配体相互结合有一定的空间结构要求,空间结构改变对效应存在一定定量关系
- 信号传导机制(通路 passway):受体药物结合后,经过通路将生物信号传递放大,最终产生可见效应
- 孤儿药和孤儿受体:没有找到内源对应受体或配基的药物和受体
- 基于假设:人类发现的药物受体,在体内一定有对应可以和药物靶点结合的内源配体
- 如阿片(吗啡)受体,药物吗啡可以作用于该受体,对应内源物质为内啡肽(内源性镇痛系统)
-
占领学说
- 定义:首先药物和受体结合形成复合物,复合物再通过一系列信号转导,发挥效能
- 特性
- 符合质量作用定律
- 形成复合物为可逆过程
- 药物浓度和药效的关系
- 复合物和药效成正比,受体被药物占领得越多,药效越来越强
- 若药物浓度远远大于kd,药效无限接近于100%最大效能
- 若药物浓度正好等于kd时,药物正好产生50%(kd可用ec50替代)
- 测量实验方法
- 测量当药物效能发挥达到一半时的药物浓度(直接测量ec50)——寻找量效曲线
- 用同位素标记法标记配体或者受体,测量药物和受体的结合率
理论上测量的EC50和药物受体结合率相等
-
亲和力和内在活性
- 亲和力 Affinity
- 由kd的大小反应(和亲和力成反比),为kd的导数
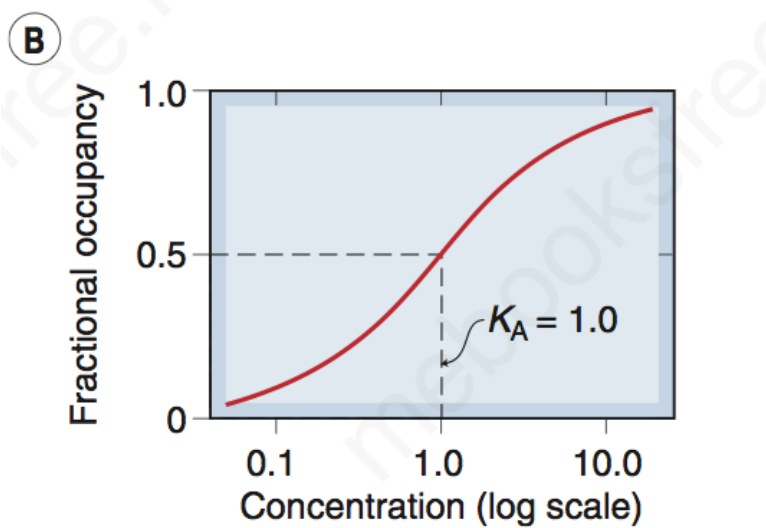
- 受体被占据一半时,ka等于此时药物浓度
注意EC50和Ka只是有线性相关关系,并不完全等于ka
- 内在活性 Intrinsic Activity α
- 反映配基激活受体之后产生药效能力

范围在0-1之间 - α为0表明没法引起任何生物学效应,等于1能产生最大的生物学效应
- 反映配基激活受体之后产生药效能力
- 亲和力 Affinity
-
药物分类
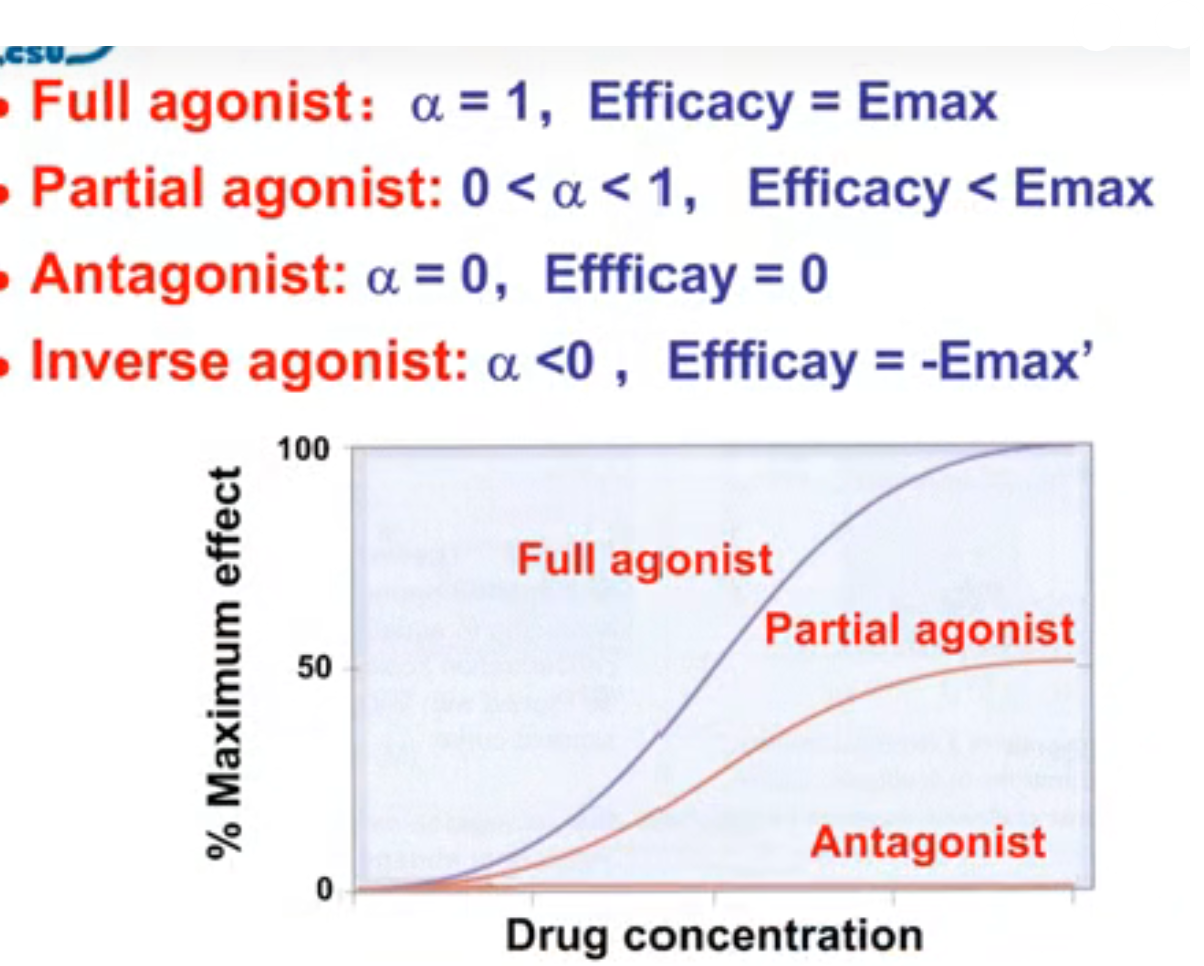
- 激动剂 和靶点结合会引起最大效应
- 拮抗剂/受体阻断剂 有亲和力但没有活性,封闭受体阻断激动剂作用,不引起任何效应
- 部分激动剂 效应介于上述两者之间,比激动剂弱
- 反向激动剂 引起和激动剂反向的效应
阻断剂一般分为竞争性和非竞争性阻断剂
-
拮抗剂分类
-
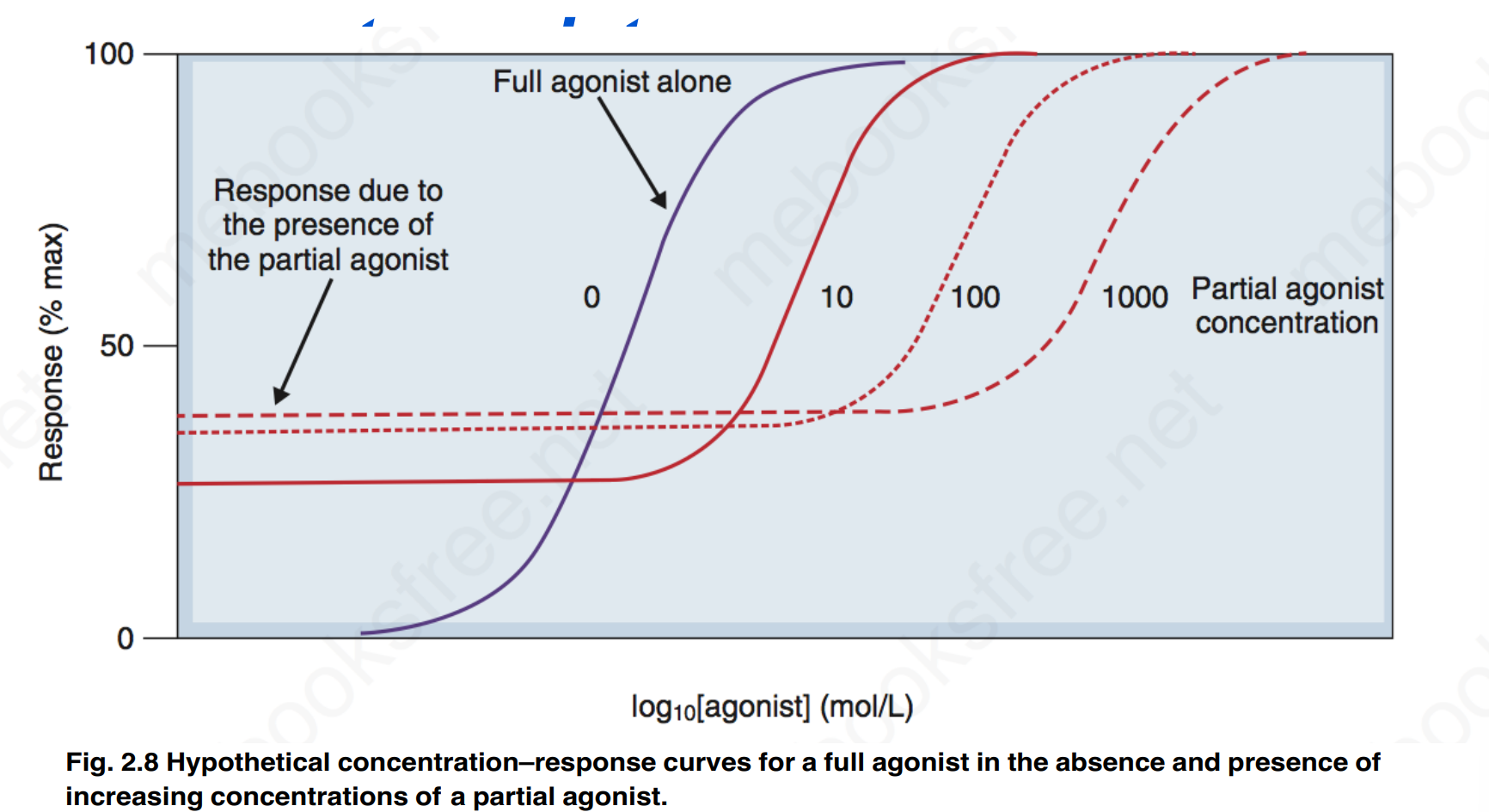
竞争性阻断剂:和激动剂竞争结合相同位点,效果取决于浓度比(给kd×系数,实则增大kd降低亲和力)
- 具体还可分为可逆性和非可逆性

造成s型曲线的平行右移的原因:
- 相同药物比,使用竞争性阻断剂
- 不同药物比,几种药物的ec50(kd)不同
- 具体还可分为可逆性和非可逆性
-
非竞争性阻断剂:作用在同一个受体但和激动剂位点不同,不会和激动剂相互竞争,这样能造成受体分子构型改变,达到降低活性的目的

> 为什么ec50多数时候小于kd
> 存在储备受体,不需要占领所有受体就能达到最大效应
> 生物学意义:提高系统反应的灵敏性,保持系统稳定性别构效应的影响:
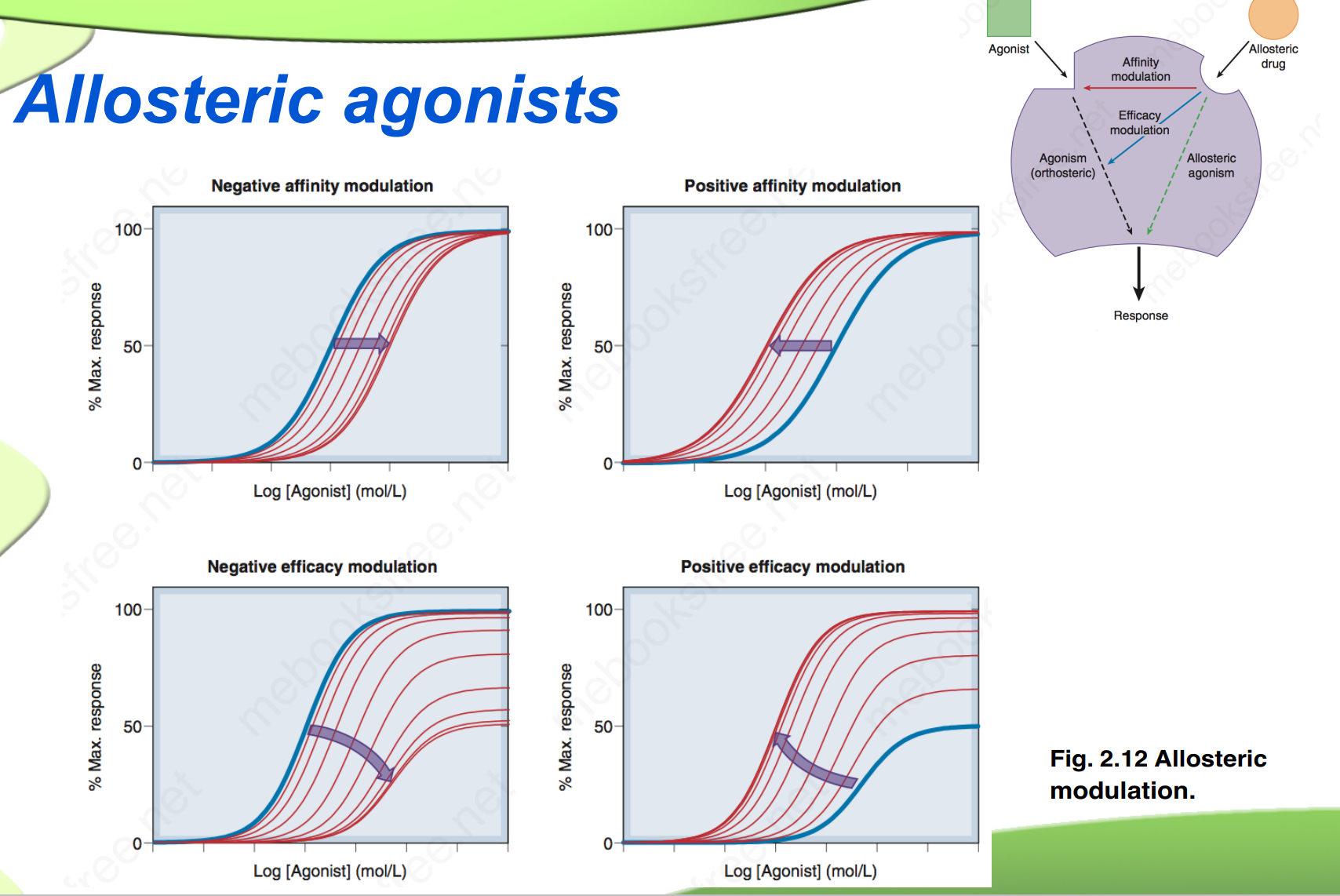
反向激动剂的影响:
-
-
受体类型分类
- 离子通道偶联受体(channel linked receptors)
- 如:神经递质传递,乙酰胆碱N受体和钠离子通道偶联
- G蛋白偶联受体(G-protein coupled receptors)
- 如:肾上腺素和β受体结合
- 酶联受体(Enzyme linked receptors):通过酶活性改变,引起蛋白质的磷酸化和去磷酸化,造成信号转导
- 如:胰岛素和胰岛素受体结合,引起酪氨酸激酶磷酸化
- 细胞内受体(Intracellular receptors)
> 并非所有受体都定位在膜表面
> 胞内受体的配体相对具有亲脂,更容易进入胞内- 如:糖皮质激素(类固醇激素都可)和对应胞内受体结合,复合物进入细胞核调控转录单元表达功能蛋白产生效应
- 离子通道偶联受体(channel linked receptors)
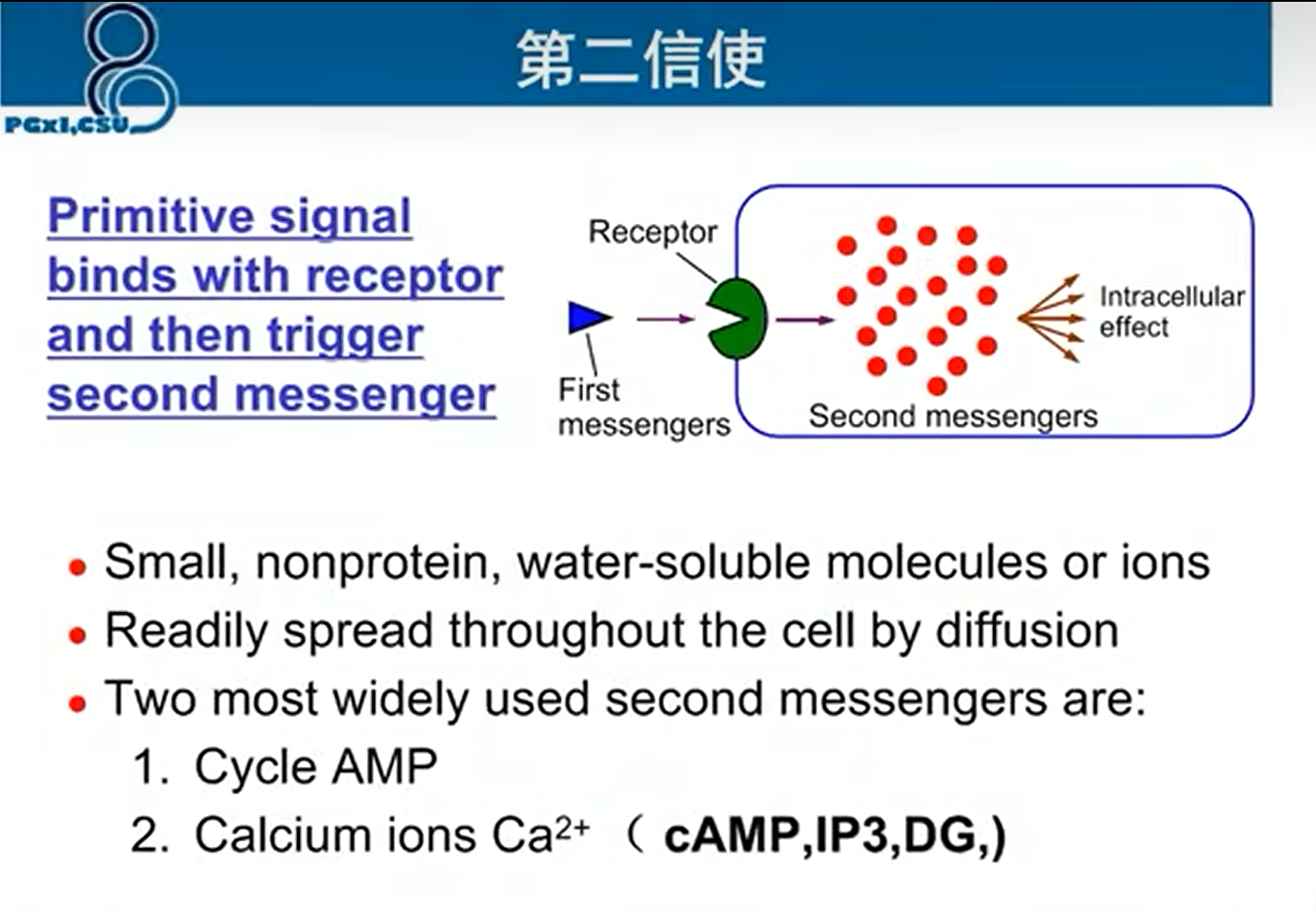
胞内信号转导 第二信使
药物作用在受体产生对应调控信号之后就会失去活性,被代谢降解;信号进入细胞后由第二信使完成信号级联放大,调控基因表达等功能
-
常见的第二信使: cAMP, cGMP,, IP3,DG
-
受体调节:与配体作用过程中相关数目和亲和力变化称为受体调节
- 上调(反跳)和下调(耐受)
- 下调:受体数目减少,相应药理性能下降。在激动剂过多的时候,相应受体表达下调,维护细胞内环境稳定
长期服用受体激动剂引起耐受,如哮喘病人常用β受体激动剂
- 上调:反之在拮抗剂过多的时候,相应受体表达上调。
长期使用β受体阻断剂治疗心绞痛,引起受体表达上调,如果突然停药会造成β受体的内源性配体——肾上腺素过于敏感,引起更强的反跳症状
- 下调:受体数目减少,相应药理性能下降。在激动剂过多的时候,相应受体表达下调,维护细胞内环境稳定
- 同种调节:配体使得自身受体变化
- 如β受体阻断剂使得β受体数量上调
- 异种调节:不是该受体的配基对该受体产生影响
- 如糖皮质激素使得β受体上调,临床中使用糖皮质激素和β受体激动剂联用,降低耐受效应
- 上调(反跳)和下调(耐受)
量效关系 Dose-effect Relationship
- 量效曲线
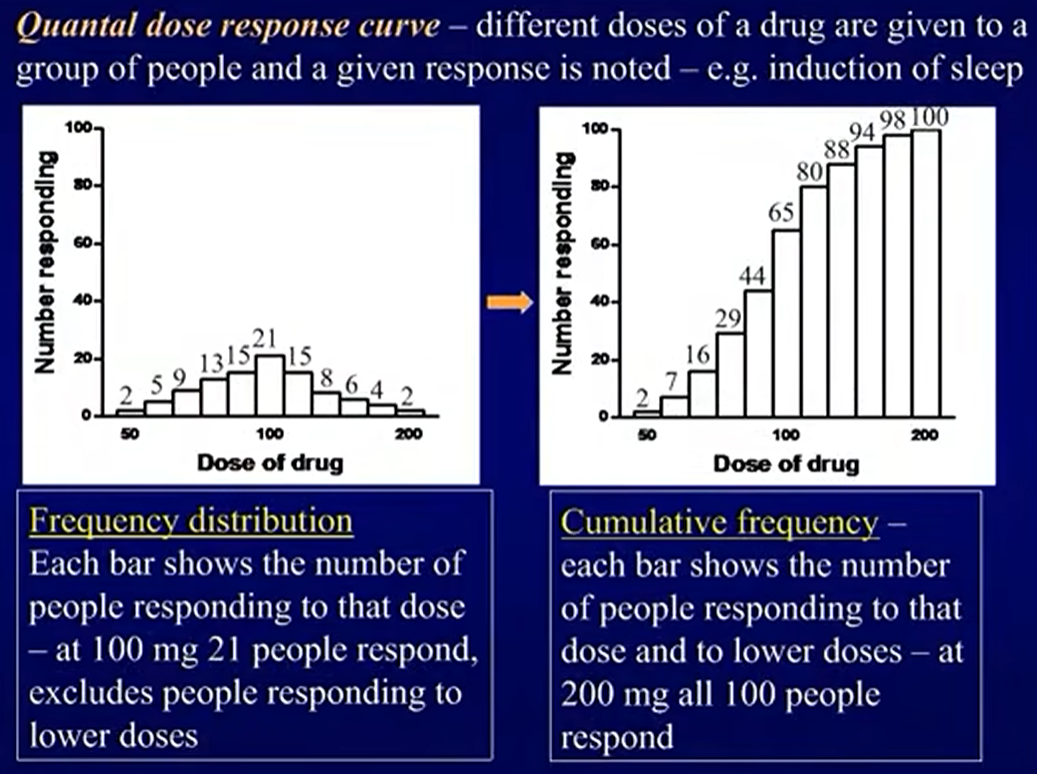
- 质反应(Qualitative effect): 以定性标准来测定药物效应的方法
- 标准如阴性阳性、有效无效
在该情形下为累计函数曲线
相关参数
ED50 是半数有效量,即所有个体敏感的最小有效量的均值
TD50 以中毒为指标就是半数中毒量,同理 LD50为半数致死量
以上三种指标用来衡量药效强弱和安全性
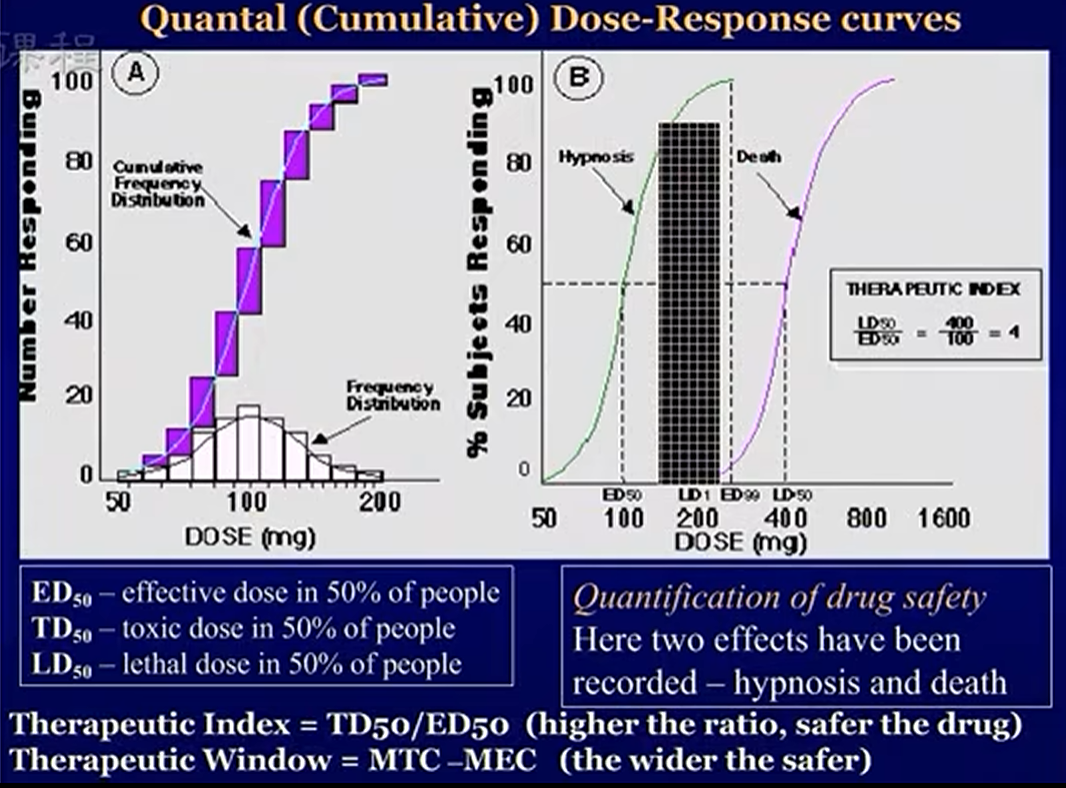
治疗指数:LD50/ED50,用于描述有效量到致死量的距离差————反应药物安全性
治疗窗口:MTC(最小中毒量)-MEC(最小有效量)。治疗时会保持血药浓度停留在治疗窗口内,但是这个剂量因人而异;敏感个体窗口整体变低,耐受个体窗口整体变高;窗口宽窄反映药物安全性
- 标准如阴性阳性、有效无效
- 几个重要概念和剂量的关系
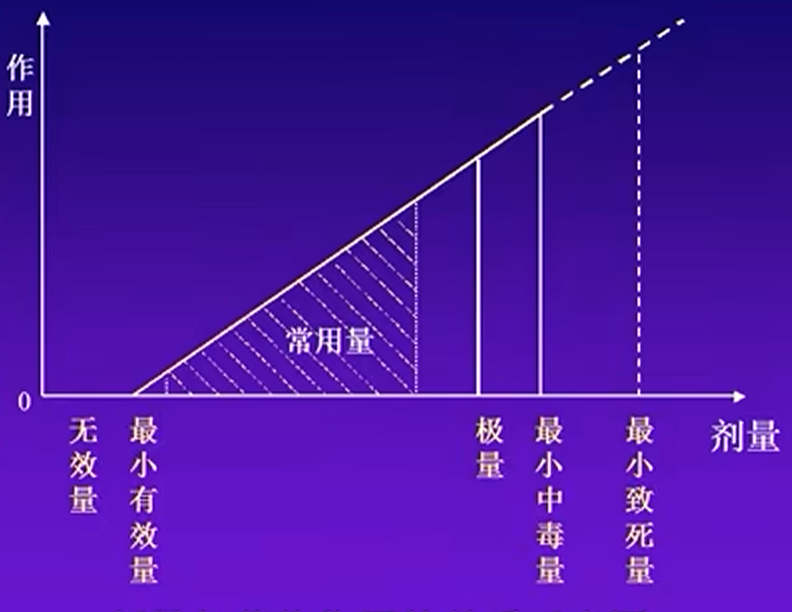
- 常用量一般高于最小有效量低于最小中毒量;在常用量中临床治疗的最大剂量为极量
> 也有情况超过剂量用药,比如激动剂中毒,使用过量拮抗剂抵消药效
- 常用量一般高于最小有效量低于最小中毒量;在常用量中临床治疗的最大剂量为极量
- 量反应(Quantitative response):以连续可数值化的标准来测定药物效应的方法
- 标准如血压、心率、血糖和酶活等
- 该情形为一个连续双曲函数,取对数后变为s型曲线
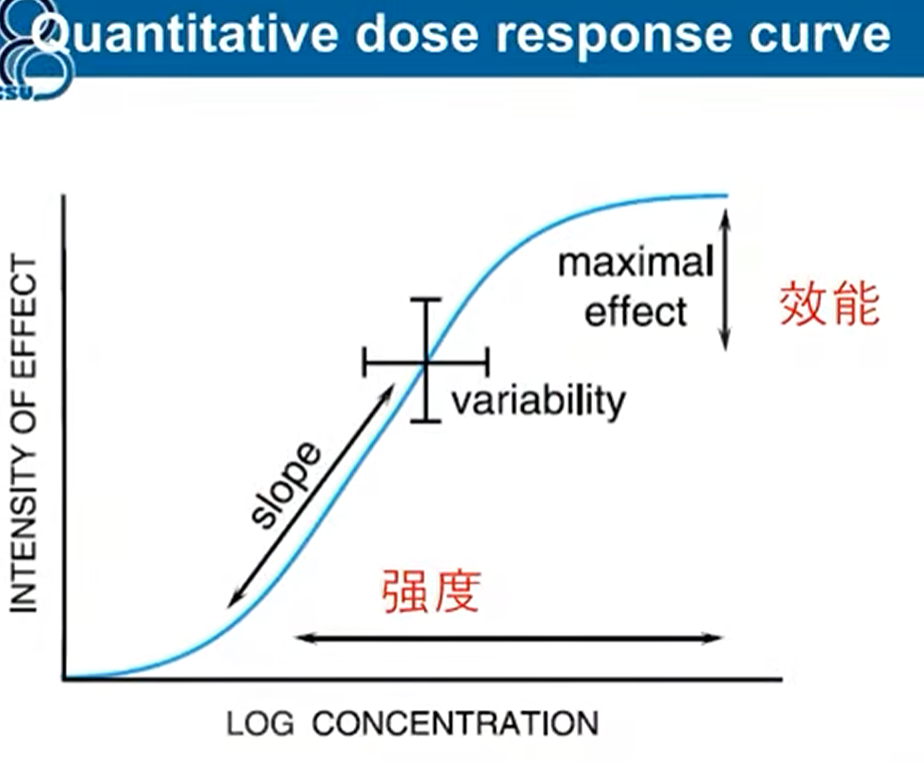
- 相关参数
- 效能(efficacy):药物能产生的最大效应
- 强度(potency):产生某一效应时的相对剂量;药物强度越大达到相同药效所需剂量越小
- 效能和强度的比较
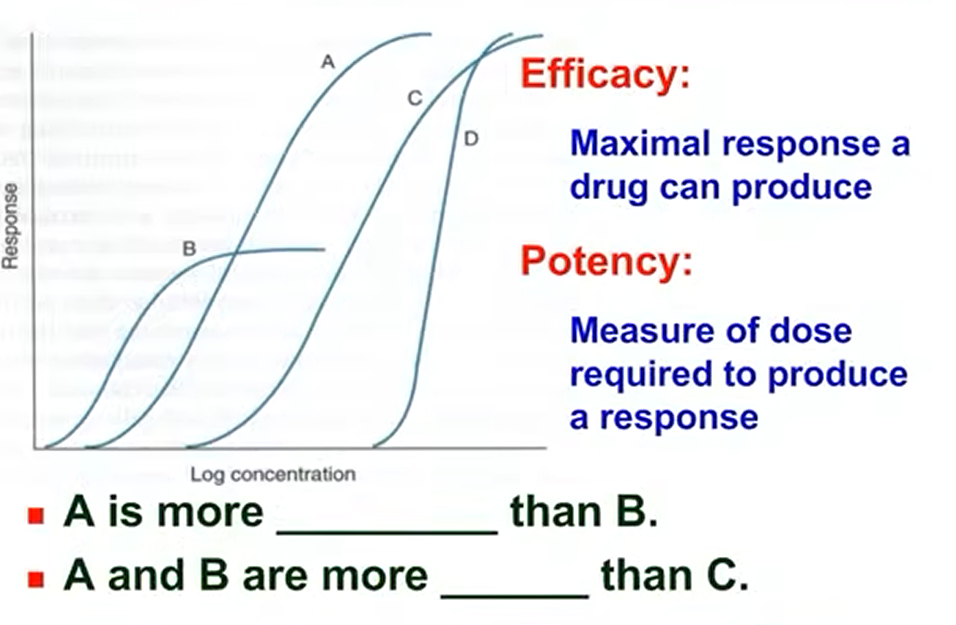
> 在临床上效能强的药物意义更大 - 效能和效价强度的关系
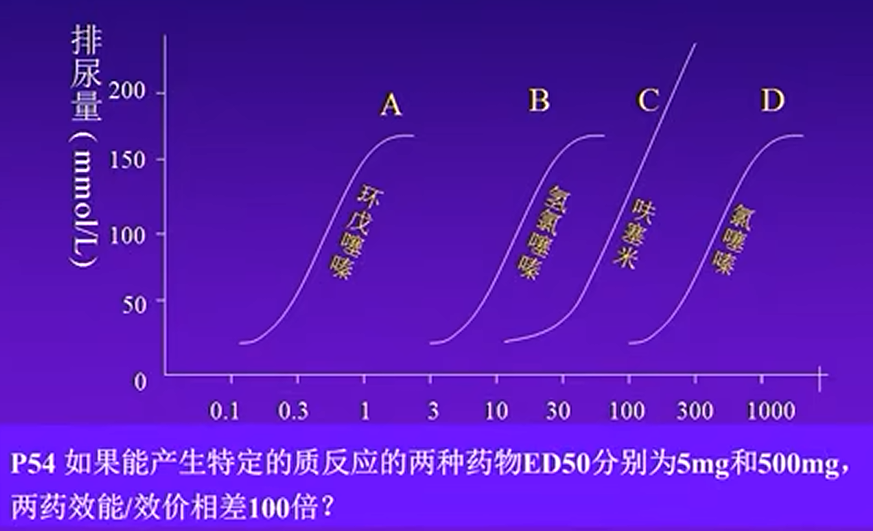
- 思考题
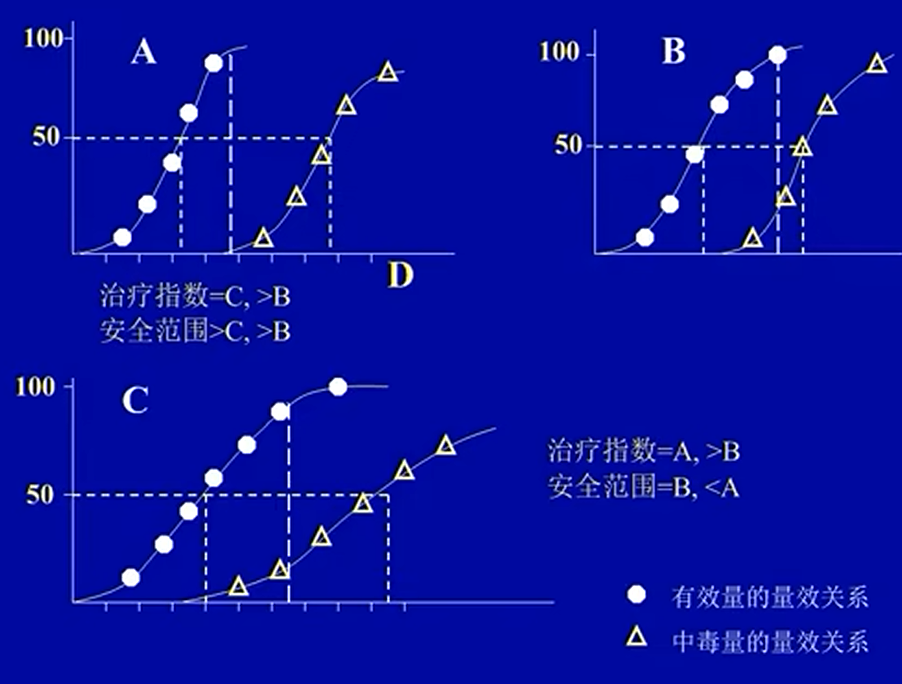
- A比C安全范围大,因为在C的高有效剂量下,也达到了部分的中毒剂量,但是A达到最大药效时的最小剂量仍比中毒最小剂量低。
- B比a治疗指数小,两条线间距更近
- 质反应(Qualitative effect): 以定性标准来测定药物效应的方法
药物作用机制 Mechanisms of Drg Action
- 非特异性药物作用机制
- 与药物理化性质相关
- 和药物(立体)化学结构关系不大
- 特异性药物作用机制
- 药物生物活性与化学结构紧密相关
- 药物作用特异性靶点
- 影响药物作用的因素
- 耐受性 vs 耐药性
- 耐受性(tolerance):机体多次用药后对药物反应下降
- 耐药性(resistance):病原体或肿瘤对药物的敏感性下降
- 躯体依赖 vs 精神依赖
- 共同表现是用药个体有再次用药的强烈意愿
- 区分标准是停药之后是否出现戒断症状
- 药物相互作用
- 相加:两种药效叠加
- 增强:两种有效药物相互促进发挥作用
- 增敏:自身无药效的药物增强了另外一种药物的效果
- 拮抗:两种药物联用药效减弱,或无直接药效的药物降低了另外一种药物的效果
- 安慰剂(Placebo):不含活性药物但是又暗示某种效应的制剂(在外形、颜色、味道等方面与某种活性药物相同)
- 耐受性 vs 耐药性
传出神经系统药理
神经系统概念
-
神经系统结构
- 传出神经
- 自主神经系统(植物神经系统)
- 运动神经系统
- 传入神经————感觉神经系统
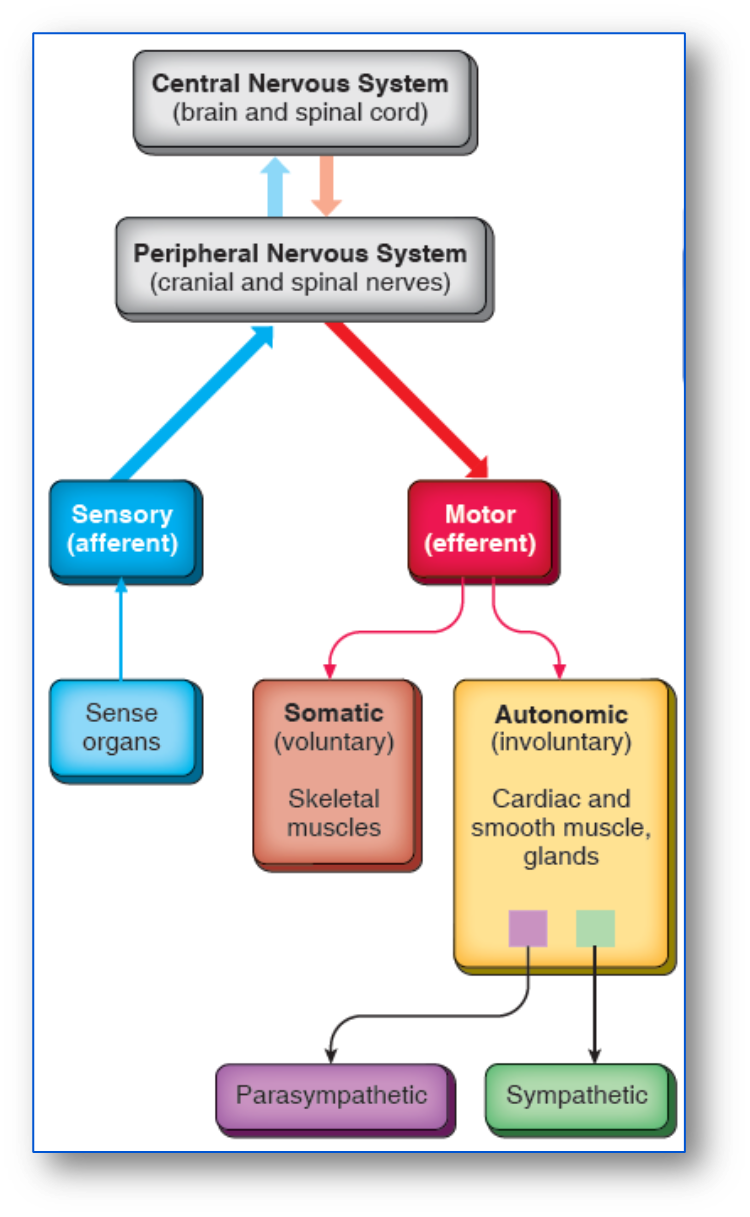
- 传出神经
-
传出神经分类
- 按解剖分类
- 交感神经
- 副交感神经
- 运动神经
- 按递质分类
- 胆碱能神经
- 全部交感/副交感N节前纤维(支配肾上腺髓质的交感神经)
- 副交感神经的节后纤维
- 运动神经
- 极少数交感N节后纤维(汗腺、骨骼肌血管舒张N)
- 去甲肾上腺素能神经
- 几乎所有交感N节后纤维(去除上述少数)
- 多巴胺能神经
- 支配肾脏血管的交感神经节后纤维
- NANC能神经(多类)
- 肠神经系统的某些神经纤维
- 胆碱能神经
- 按解剖分类
-
植物神经解剖分类(按神经递质)
- 植物神经节前纤维递质:均为Ach
- 交感/副交感
若大量乙酰胆碱释放,会首先引起神经节兴奋,使得交感和副交感同时兴奋,生理情况不容易出现
- 交感/副交感
- 植物神经节后纤维递质:不同
- 副交感神经:均为Ach
- 交感神经:主要为NA,例外:
- 汗腺N/骨骼肌血管舒张N——Ach
- 肾脏血管——DA(多巴胺)
- 植物神经节前纤维递质:均为Ach
很多药物通过影响这些递质和受体进行调控发挥作用
-
神经系统解剖结构
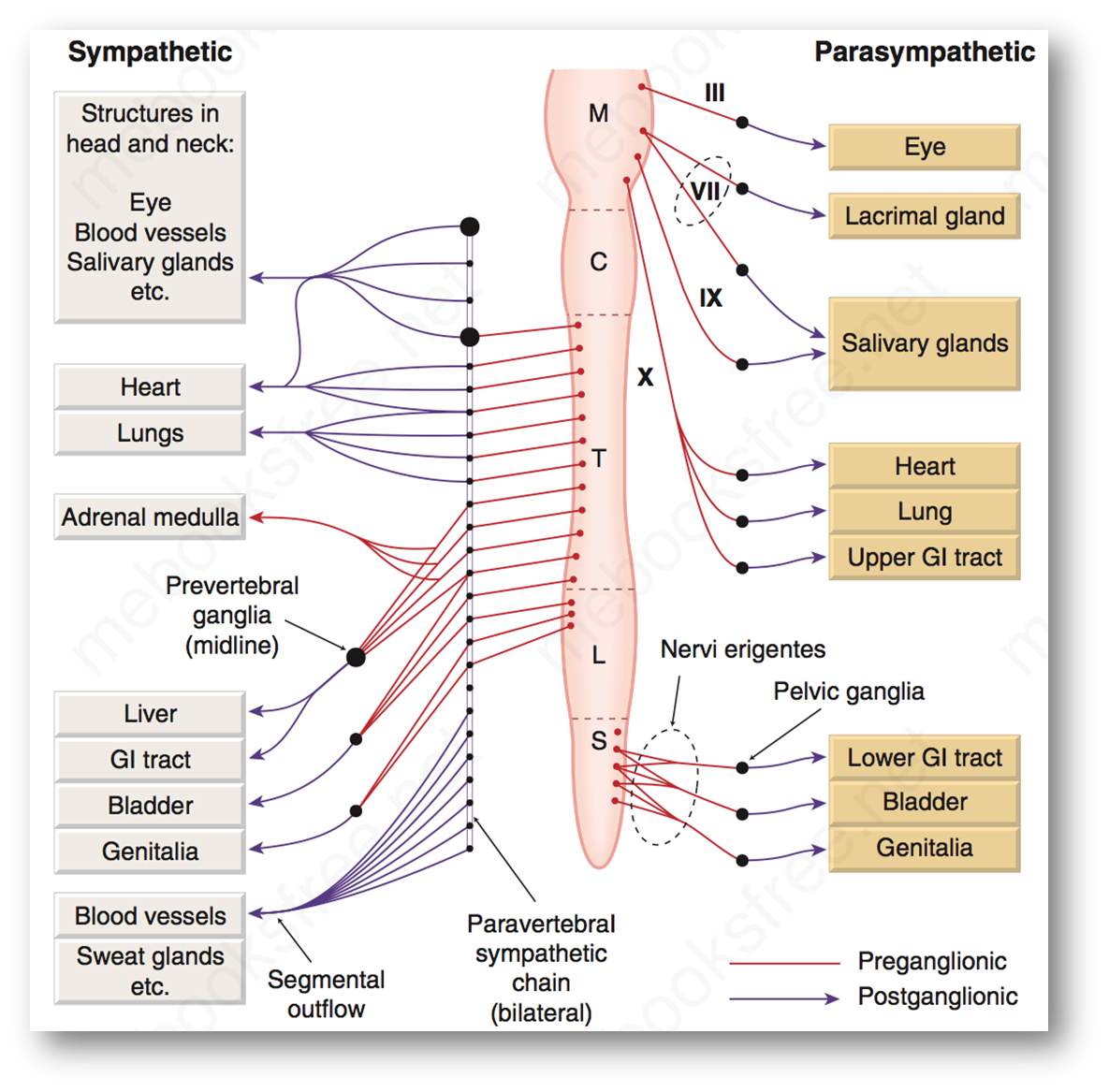
-
神经节是功能相同的神经元在中枢以外的周围部位集合而成的结节状构造
-
自主神经周围传出纤维的交感部,即交感神经。它的组成也包括节前神经元、节前纤维和节后神经元、节后纤维。由纤维组成交感神经、神经丛,由节后神经元组成交感神经.
-
交感神经 (sympathetic division)支配应对紧急情况的反应;副交感神经 (parasympathetic division)监测身体内部功能的常规运行。
基本区别:
- 位置不同:交感神经位于脊髓胸段及以上,副交感神经则起源于脑干及脊髓的骶部或颈部。
- 神经纤维长度不同:交感神经纤维很短,神经节位于脊柱旁;而副交感神经纤维较长,神经节常常位于内脏器官附近。
- 鉴别标志不同:交感神经的鉴别标志是前根短、后根长,背根和前根合成交感神经干;副交感神经的鉴别标志是前根长、后根短,背根与前根终止单独形成副交感神经干。
- 发射神经递质不同:交感神经引导的神经递质为肾上腺素、去甲肾上腺素等,能够使身体进入应激状态;副交感神经引导的神经递质为乙酰胆碱,使身体保持安静和放松状态。
- 生理作用不同:交感神经主要调节体内代谢、心率、呼吸、血压等处于应急状态下的生理反应;而副交感神经则主要调节消化、泌尿、生殖器官等处于休息状态下的生理活动,有利于能量储存和恢复。
- 一个人紧张之后冒汗是交感神经兴奋的结果,但是其递质不是去甲状腺素而是乙酰胆碱
- 在交感神经中有两处释放的不是去甲状腺素:一处是汗腺(乙酰胆碱),另一处是肾脏血管(多巴胺)
- 肾上腺髓质可以看成大交感神经节(递质和受体和神经节一样),只受到节前纤维调节(其上有N受体),并直接向血液中释放肾上腺素或去肾上腺素
-
-
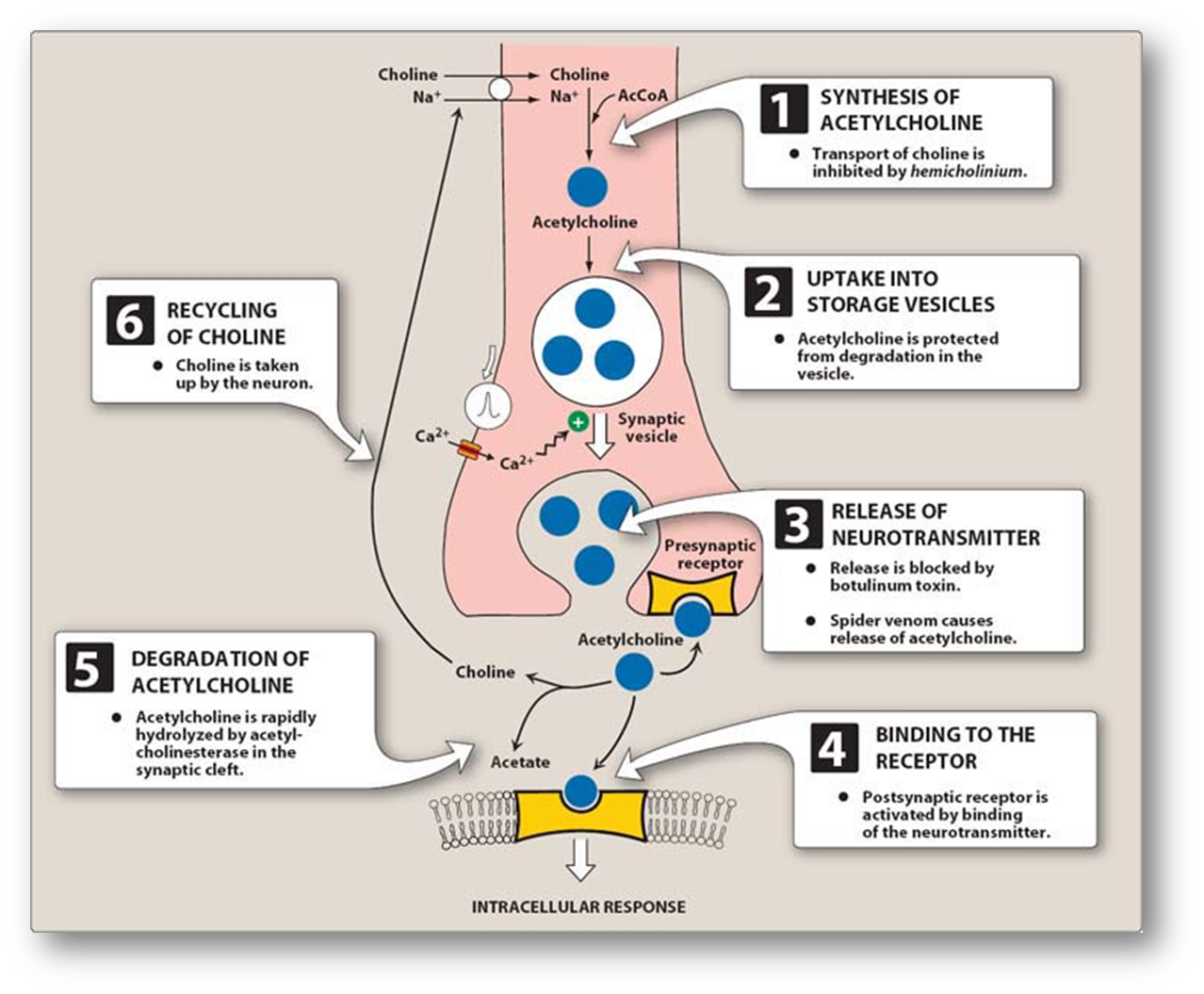
传出神经系统的递质
- 递质(合成限速酶/作用终止主要方式)
- Ach: 胆碱乙酰化酶/胆碱酯酶水解(效率极高)
- NA: 酪氨酸羟化酶/摄取(1-再利用,2-酶解)
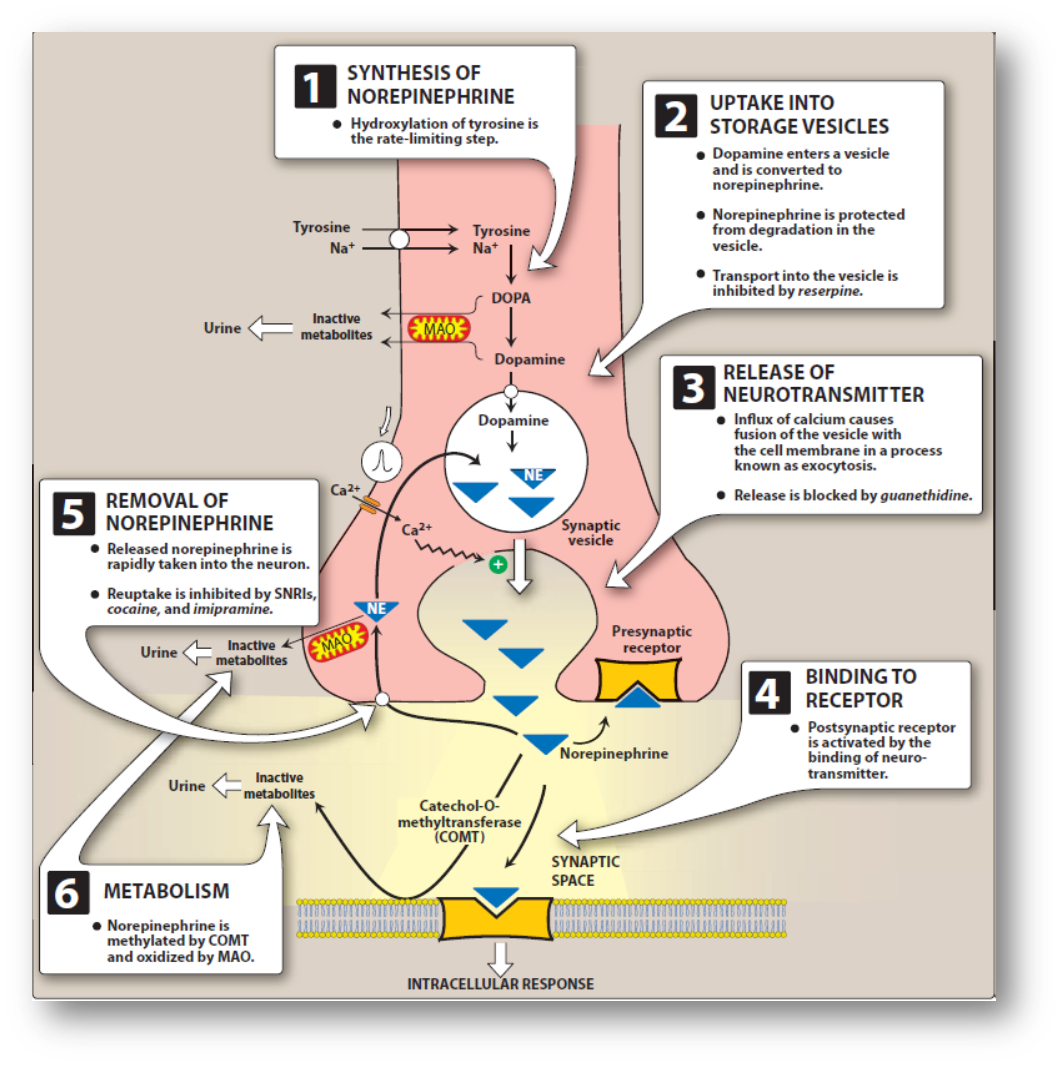
- NA合成代谢步骤:
- 合成: 酪氨酸->多巴->多巴胺-多巴胺β-羟化酶(DβH)->NA
- 囊泡:转运->摄取->合成->贮存
- 胞吐外排:神经冲动去极化,Ca2+内流(N型钙通道,阻滞剂常见L型钙通道),使得囊泡和突出前膜融合
- 摄取1:突触前膜重摄取(>75%),大部分再利用,小部分被MAO酶降解
- 摄取2:非神经组织重摄取(少量),被COMT,MAO破坏
- 扩散:入血->肝,肾,被COMT、MAO破坏(通过测血浆中NA的浓度反应交感神经的张力)
- 如果抑制NA的再摄取过程,会造成受体短时间堆积(局部停留时间变长)先兴奋,长时间由于神经递质在体循环内被快速降解(快于合成),引起神经细胞耗尽递质NA,进而交感神经功能受到抑制
- 抑制NA降解酶的效果远不如抑制Ach降解酶对交感神经功能的抑制效果好
- 分解:单胺氧化酶(Monoamine oxidase)
- Ach合成代谢步骤
- 合成:胆碱乙酰化酶
- 运输:胞吐
- 分解: 胆碱酯酶
- Ach: 胆碱乙酰化酶/胆碱酯酶水解(效率极高)
- 递质(合成限速酶/作用终止主要方式)
-
受体的生物效应
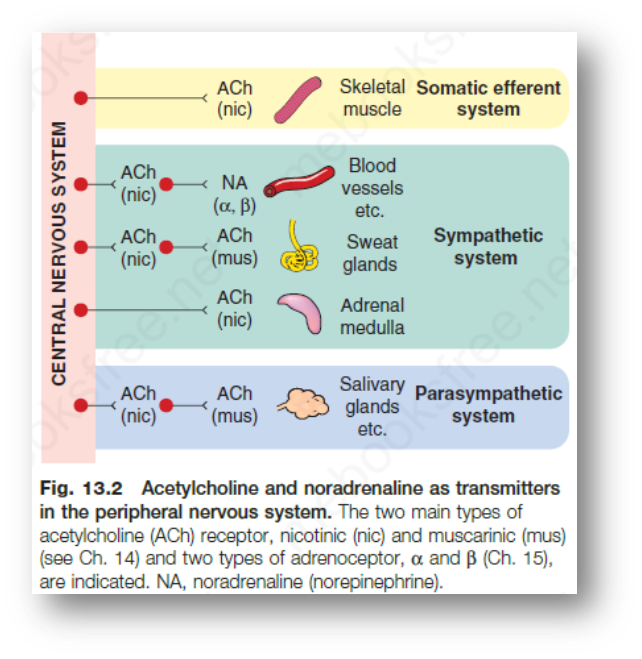
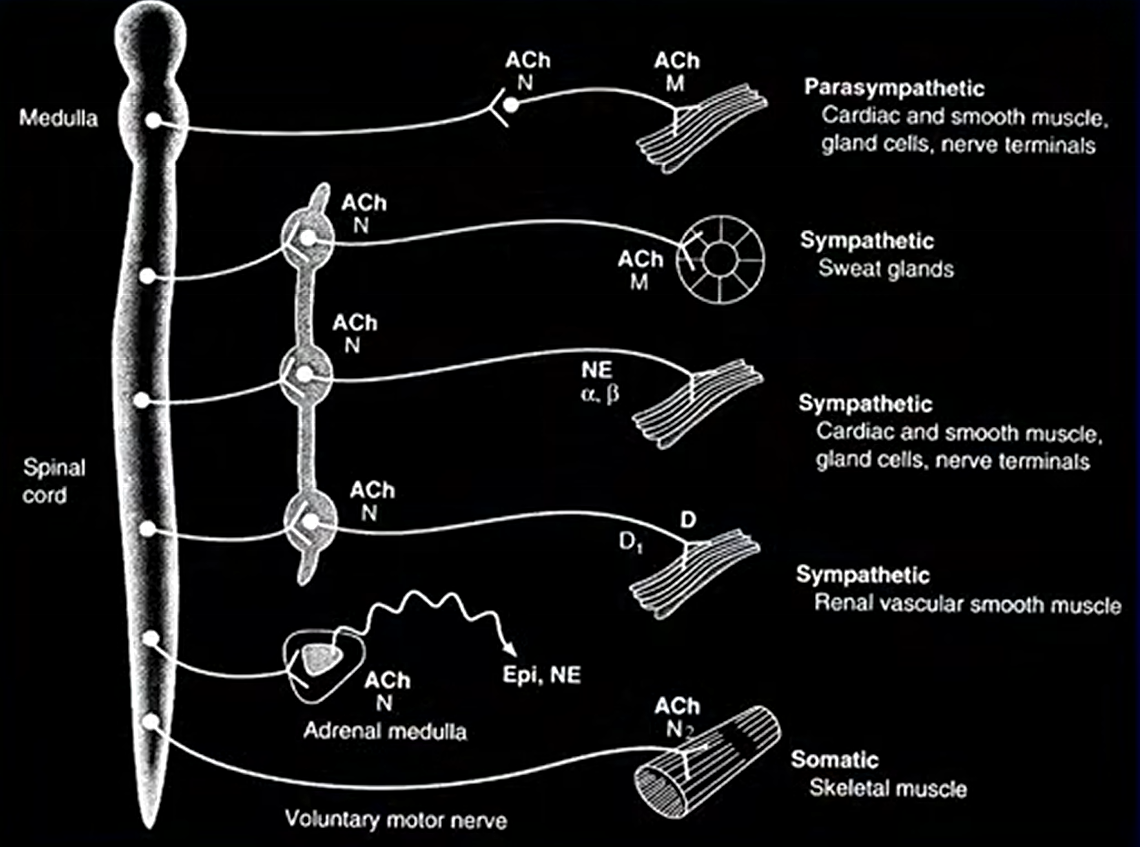
-
运动神经:骨骼肌收缩(Ach,NM受体)
-
交感:运动、应激————主要影响心血管系统
- 运动情形分析
- 运动时,心脏血管相关交感神经兴奋,心跳加快血管扩张,但内脏皮肤粘膜相关血管平滑肌收缩,以实现血流再分配,增加骨骼肌的血流量,所以骨骼肌血管扩张
- 内脏皮肤粘膜血管的收缩是依赖α受体控调控,而骨骼肌血管的扩张是依赖NM受体(胆碱类神经)调控
- 对氧需求量增多,呼吸道交感神经兴奋,使得呼吸道平滑肌舒张使得支气管扩张,有利于通气
- 运动情形分析
-
副交感:休整、积蓄能量
-
双重支配 -> 优势效应
- 交感优势:心血管
- 副交感优势:消化、泌尿、腺体、眼
- 当交感/副交感神经共同控制器官时,根据优势地位确定主要控制方向
- 如:心脏,交感神经占据的部分比副交感神经更多,所以以交感神经为主;若使用神经节受体激动剂,会使得交感神经兴奋从而使得血压上升;若使用神经节兴奋阻断剂,会使得交感和副交感的效果都被抑制,只体现出优势神经的支配效果,心血管系统的反应是血压下降,是临床曾经使用过的紧急降压药物(现已弃用,选择性差)
-
-
胆碱受体
M型区分不严格,现有临床抗胆碱药物没有太多选择性
- M型5种亚型(Muscarinic 毒蕈型 G蛋白偶联受体)
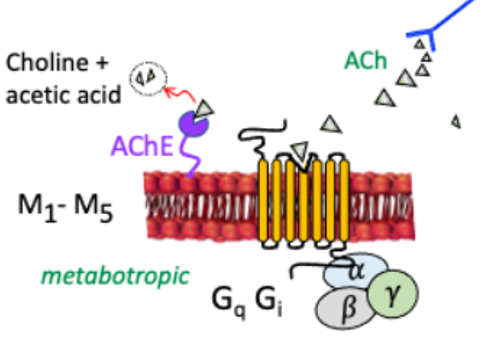
- :胃壁细胞
> 哌仑西平,Pirenzepine:该抗胆碱药物对胃壁细胞的毒蕈碱受体有高度亲和力。然而临床上用于减少胃酸分泌效果有限,原因是乙酰胆碱类神经对胃酸分泌贡献少,主要是组胺 - :心肌、内脏(非血管)平滑肌
- :腺体、血管平滑肌(西维美林,Cewimeline)
- 、:CNS
- :胃壁细胞
- N型2种亚型(Nicotinic 烟碱型 配体门控离子通道)
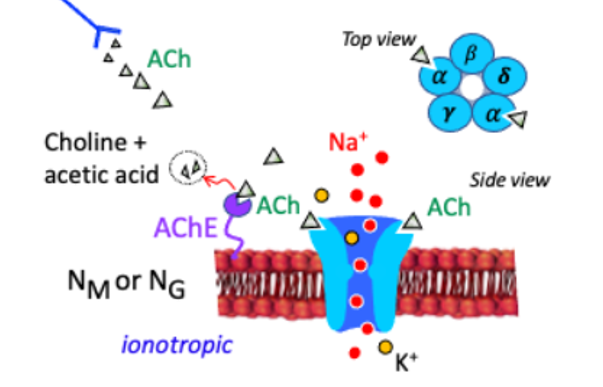
- :神经节
- :骨骼肌
实例分析:
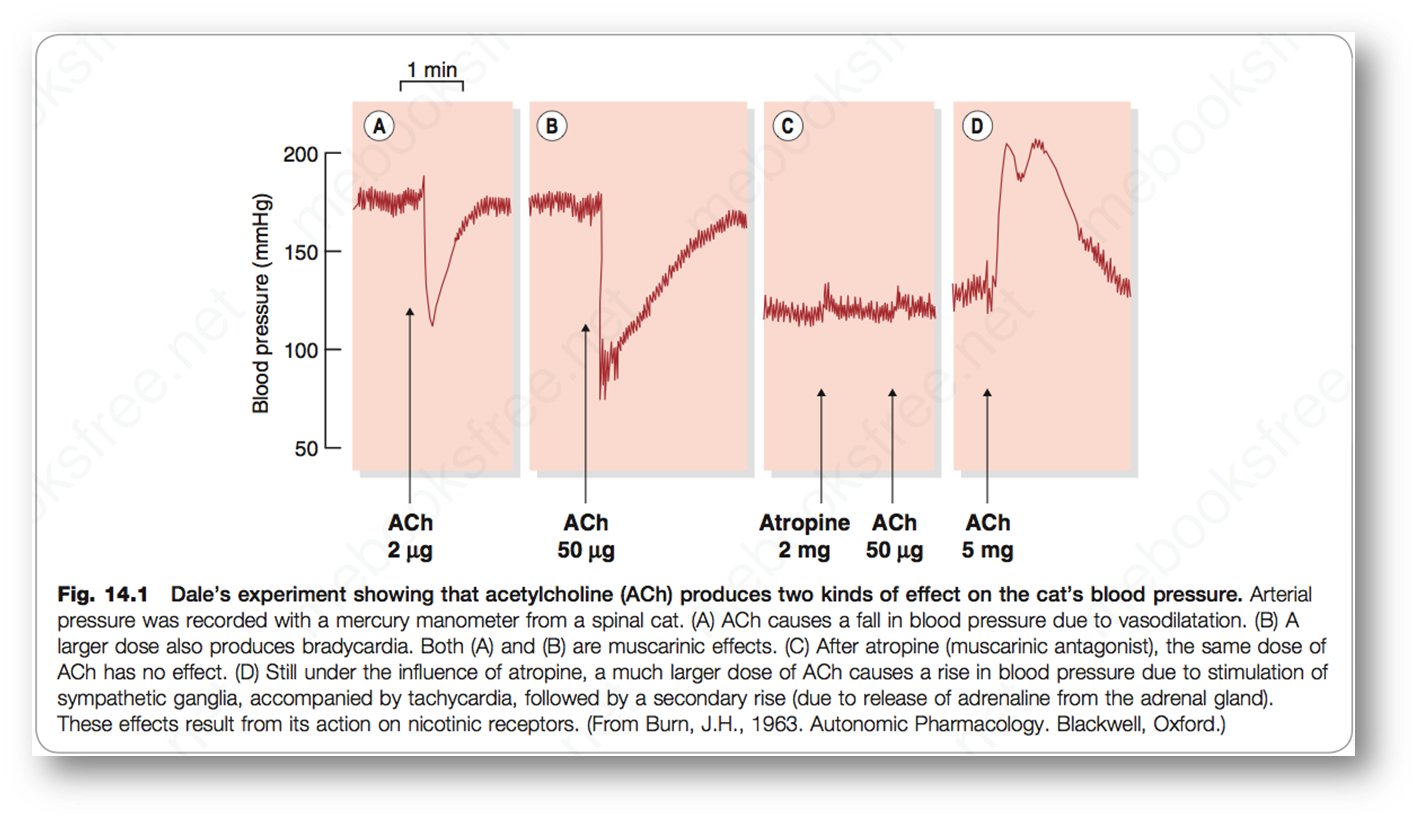
- AB图说明在较低浓度下ACh激动M受体使得副交感神经兴奋,引起血压下降
- C图说明在M型竞争型胆碱受体抑制剂阿托品的作用下,ACh的效果被抑制,不引起变化
- D图说明在高浓度ACh的刺激下,ACh对N和M受体共同产生作用,同时因为浓度比较高,阿托品的竞争效果也随之减弱(解释中间为什么有下降),当ACh大量激动N受体(神经节)引起了肾上腺素能神经(交感神经)的兴奋,造成血压上升。
- D图可以说明M受体对ACh亲和力比N受体高很多
- M型5种亚型(Muscarinic 毒蕈型 G蛋白偶联受体)
-
肾上腺素受体
现有的肾上腺素α受体阻断剂,抑制α1型受体
生物学本质(微观原理):
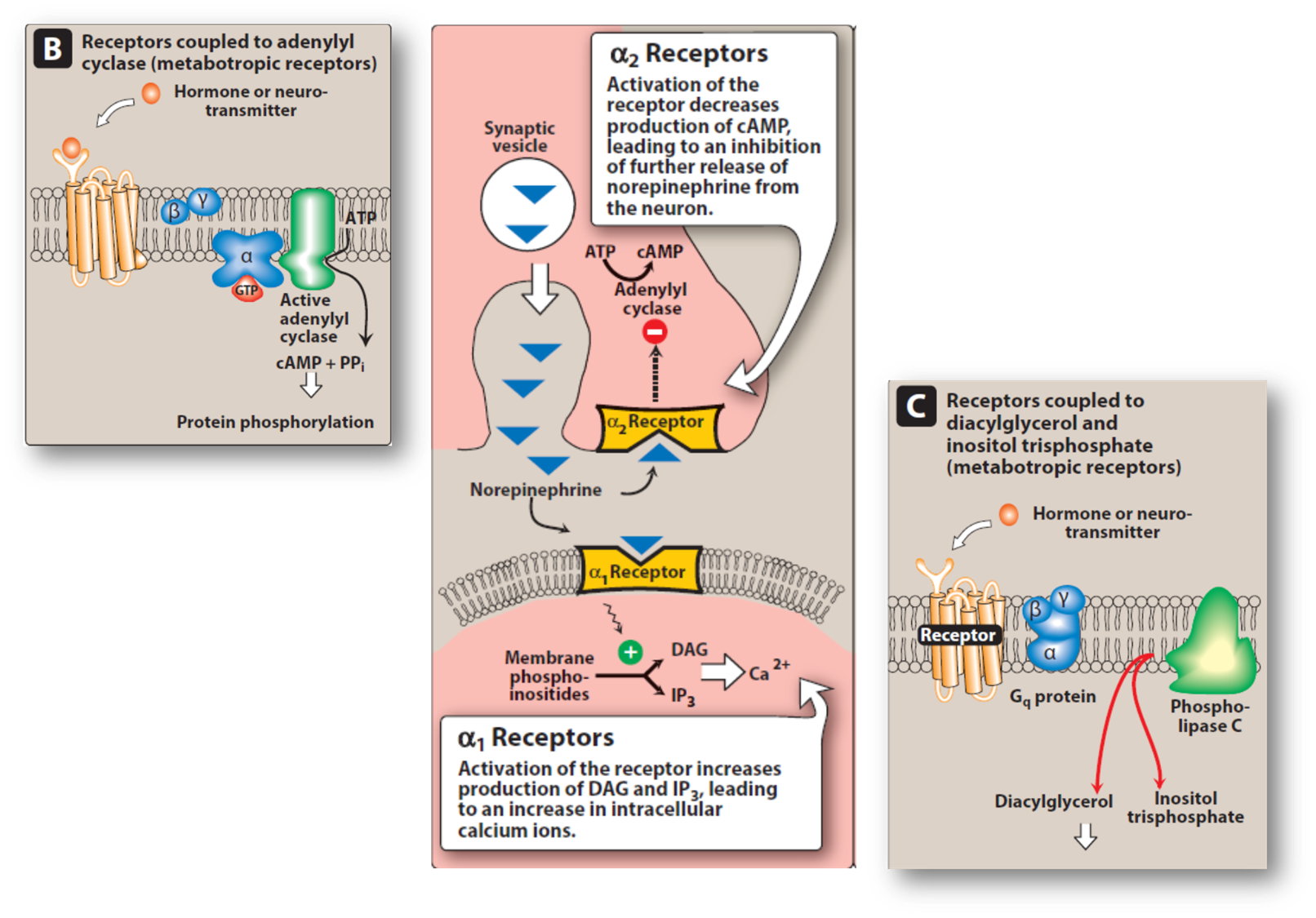
-
平滑肌收缩:缩血管、扩瞳孔、抑排尿
-
外周——交感N突触前膜:抑制NA释放
中枢——交感N突触后膜:交感中枢 -
:心肌兴奋
-
: 支气管平滑肌(扩张)、血管平滑肌(舒张)、骨骼肌(颤动)、糖原分解
-
:脂肪分解
总结
-
-
多巴胺受体(外周)
- 肾血管平滑肌(舒张)
- 突触前膜、平滑肌
药物作用方式
- 作用于受体:激动剂、拮抗剂(阻断药)
- 影响递质
- 合成、转化(影响胆碱能神经):合成(工具药)、酶解
- 贮存、释放(影响去肾上腺素能神经):干扰再摄取(利血平)、促释(麻黄碱)
分类
- 胆碱能神经——影响降解
- 拟胆碱药: 胆碱受体激动药、抗胆碱酯酶药
- 抗胆碱药: 受体阻断药、胆碱酯酶复活药
- 去肾上腺素能神经——影响回摄
- 拟似药:肾上腺素受体激动药、NA促释药
- 拮抗药:肾上腺素受体阻断药、抗去肾上腺素能神经药(利血平)
药物对胆碱能神经元信号传递的作用
拟胆碱药
-
胆碱受体激动药
- 胆碱酯类(M、N均作用)————拟胆碱药:其为包括乙酰胆碱在内的数个结构和功能与乙酰胆碱非常一致的药物,比如醋甲胆碱、卡巴胆碱
- 生物碱类(M样作用 副交感兴奋表现)————拟副交感神经药: 其为人工合成的能够对ACh的M受体(毒蕈碱样受体)和N受体(烟碱样受体)起到激动作用的物质,比如毛果芸香碱、毒蕈碱和槟榔碱
- 作用效果
- 心血管系统抑制(心缩力下降心率脉搏下降)
- 消化泌尿气道平滑肌兴奋
- 过度痉挛(推断不良反应):上吐下泻,排尿疼痛,呼吸困难
- 腺体分泌增加
- 唾液汗液大量分泌
- 痰分泌过多,加之气道缩小可能造成窒息
- 眼:缩瞳、调节痉挛(近视)
- 代表药物及药理
匹鲁卡品 pilocarpine(毛果芸香碱)药理- 药效原理
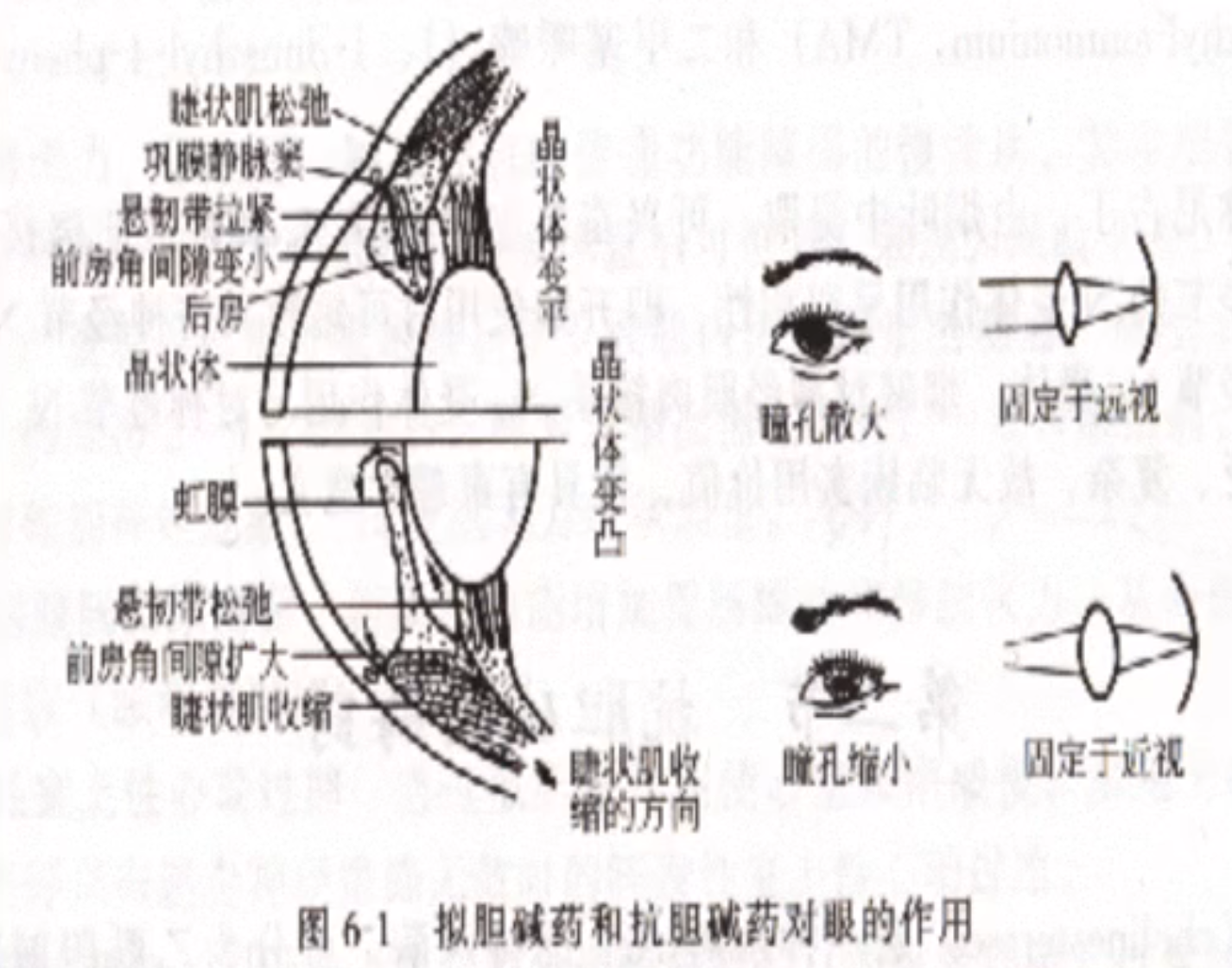
- 对睫状肌的影响
- 虹膜上有括约肌,括约肌收缩瞳孔缩小;辐射状肌(α受体支配)兴奋瞳孔散大
- 睫状体上有环形睫状肌(m受体)收缩后睫状体变小
- 晶状体环韧带拉住晶状体,当睫状肌收缩,肌环变小晶状体韧带变松弛,晶状体由于自己弹性变秃,折光率变大,引起调节近视;当睫状肌舒张,肌环周长变大,进而悬韧带拉紧,晶状体凸度下降,引起调节远视。
- 神经和瞳孔调节关系:当副交感节后纤维兴奋时,会激动M受体,造成睫状肌收缩;当交感节后纤维兴奋时,会激动β受体,导致睫状肌舒张。
- 睫状肌收缩过度————调节痉挛(引起假性近视)
- 对虹膜的影响
- 虹膜内有环绕瞳孔排列的平滑肌,叫缩瞳肌(瞳孔括约肌),以及自瞳孔周围呈放射状排列的平滑肌,叫散瞳肌(瞳孔开大肌)。前者受副交感神经支配,后者受交感神经支配
- 当瞳孔开大时,虹膜变大和晶状体接触面积变小
- 如果虹膜发炎时,晶状体和虹膜的大接触面积会由于炎症分泌物(纤维蛋白)的大量分泌可能引起粘连,造成晶状体不能形变
- 可使用散瞳药(M受体)和缩瞳药交替使用来避免
- 对眼压的影响
- 前房角处有巩膜静脉窦;从后房睫状体和虹膜的脉络丛中滤出不含蛋白质的液体,即房水,再从后房经过瞳孔流到前房,进入前房角的巩膜静脉窦实现房水循环
- 房水充满眼睛产生的压力为眼压
- 对于病理性眼压过高,由于巩膜相对坚硬,房水的压力会影响眼球后端的视盘(没有巩膜保护),长时间压迫造成视神经乳头萎缩,先视野缩小,最后出现失明————即青光眼。
- 主要治疗方法为降低眼内压,其中一种为尽量缩小瞳孔(虹膜变大),肌肉舒张,使得前房角开大利于房水通过巩膜静脉窦流出
- 对睫状肌的影响
- 药物效果
- 眼:缩瞳、降眼内压、调节痉挛
- 腺体:汗腺唾液腺分泌增多
- 平滑肌:消化道呼吸道平滑肌收缩痉挛
- 临床应用
- 青光眼(闭角型————房水流出不畅、开角型————房水生成过度)
- 虹膜炎(与扩瞳药交替使用防炎症粘连)
- 毛果芸香碱可以解救阿托品等抗胆碱药的中毒
- 药效原理
- 作用效果
- 烟碱类(N样作用):能够选择性的只激动N受体的药物
(N-Neuron) 神经节兴奋 优势受体表现
(N-Muscle) 骨骼肌收缩,算麻痹的一种,末梢兴奋,表现为肌颤
-
胆碱酯酶抑制药(间接拟胆碱药)
- 实用意义
- 胆碱受体激动药中,一般可以用来入药的毛果芸香碱只有激动M受体的效果,没有激动N受体的作用,如果患者发生的是“副交感神经张力的整体过低”,则此时实际上需要的是同时增加M受体和N受体的激动强度
- 乙酰胆碱作为药物虽然能同时刺激M和N受体,但是在体内降解过于快速;而卡巴胆碱对M的亲和性比N受体更强,也不能完全适用
- 如果我们想要真正整体提升M和N受体的激动性,从而使那些副交感张力过低的患者得到恢复,则需要另外想办法,也就是通过减小ACh的降解,从而使得ACh的量发生堆积,进而间接提升ACh的量和效能。
- 通识特性
- 与酶的亲和力 > Ach;本身代谢慢比较难,可以阻断Ach分解
简单机理分析:
体内存在着乙酰胆碱酯酶(acetylcholinesterase),它可以快速将释入突出间隙的ACh水解掉,但是,现在如果我们将乙酰胆碱酯酶的活性抑制掉,就可以导致ACh的大量蓄积,提升了ACh的含量,从而导致副交感张力得到上升。
- 与酶的亲和力 > Ach;本身代谢慢比较难,可以阻断Ach分解
- 分类
- 可逆性抗胆碱酯酶药
- 新斯的明(全身用药的拟胆碱代表药物)————与AChE非共价键结合,形成“药物-AChE复合物”减少可以单独正常工作的AChE,从而降低对ACh的水解能力
- 难逆性抗胆碱酯酶药
- 有机磷酸酯————与AChE形成共价键结合,转化为磷酰化胆碱酯酶,从事永久失去水解ACh能力
针对上述药物的中毒,我们引入胆碱酯酶复活药,可以将难逆性抗胆碱酯酶药所抑制的AChE给重新复活,从而使得ACh堆积的现状得到改善,缓解原先的毒理学效应。
- 有机磷酸酯————与AChE形成共价键结合,转化为磷酰化胆碱酯酶,从事永久失去水解ACh能力
- 可逆性抗胆碱酯酶药
- 可逆性胆碱酯酶抑制剂
- 总体药效分析
- 与胆碱酯酶形成容易解离的非共价键,将正常的胆碱酯酶转变为几乎不能水解乙酰胆碱的“二甲胺基甲酰化AChE”。但是,因为这个“二甲胺基甲酰化AChE”是以非共价键结合出来的,所以它自己会缓慢相互解离重新形成正常的胆碱酯酶,从而胆碱酯酶的催化活性又会逐渐恢复,从而M受体和N受体的过度激动作用又会消失,患者又会恢复正常。
- 持续时间有限,不会永久降低AChE的效能,引起暂时性M受体和N受体效能增加
- 经典药物:新斯的明
- 药动学
- 吸收口服 < 注射
- 不能通过血脑屏障(季铵盐,极性强不易穿膜),角膜不透
眼科替用:毒扁豆碱(Physostigmine)和地美溴铵(Demecarium bromide)
透过BBB替用:他克林(tacrine),多奈哌齐(Donepezil),加兰他敏(Galantamine)————能透过BBB对中枢神经系统以ACh为递质的神经元的激动 - 被血浆酯酶水解
- 机制
- 与酶结合形成二甲胺基甲酰化胆碱酯酶,减少自由AChE。本质效果为ACh造成的一系列效果,通过间接抑制水解来提升ACh的含量和作用效果,不涉及到中枢
- 效能:ChE酶被抑制
- 临床应用
- 治疗重症肌无力
- 疾病原理:体内产生了大量可以特异性结合到骨骼肌接头突触后膜(也就是骨骼肌细胞膜上的N-M受体)的自身抗体,这些抗体就像一个阻断剂一样,阻止了突触后膜上的N-M受体与正常的副交感递质ACh的结合,从而使得骨骼肌没法正常被ACh所激动,从而骨骼肌收缩无力
- 治疗原理:用新斯的明抑制突触间隙的AChE,从而使得ACh无法被快速水解,进而ACh会大量蓄积,从而对冲掉ACh效能的下降,进而重新提升骨骼肌收缩力
- 非去极化型肌肉松弛药解毒
- 毒理:如筒箭毒碱是竞争性N-M受体抑制剂,会特异性结合N-M受体并阻碍其兴奋,进而导致受累肌肉发生肌无力
- 药理:新斯的明增加突触间隙ACh含量,ACh浓度增加竞争性超过筒箭毒碱,重新和N-M受体结合,恢复肌肉收缩能力
- 副作用:过量引起肌束震颤、肌张力下降(同过度激动N-M受体)
- 术后腹气胀尿液滞留(手术麻醉阻断了大部分神经)
阻断顺序(恢复反之) 先热(植物神经)、不疼、后不能动
内脏平滑肌也有胆碱类神经支配,所以手术麻醉会阻断肠道膀胱的平滑肌的M受体,引起上述问题
该药物能减慢Ach清除,所以能治疗上述症状 - 阵发性室上性心动过速
使Ach堆积,作用于心房肌的M胆碱受体,引起心率减慢
- 治疗重症肌无力
- 不良反应
- 急性胆碱能危象,大量ACh堆积,使得NM受体过度激活
- 禁用支气管哮喘————气道高反应性,Ach堆积会诱发严重的支气管痉挛
- 禁用机械性肠梗阻(堵塞)————Ach堆积加快肠蠕动,可能导致肠破裂
- 禁用尿路梗死————Ach堆积加快膀胱平滑肌收缩,可能引起膀胱破裂
- 药动学
- 总体药效分析
- 难逆性胆碱酯酶抑制剂
- 总体药效分析
- 与胆碱酯酶形成牢固结合不易解离的共价键,从而将正常的胆碱酯酶转化为磷酰化AChE,不具有水解ACh的能力。
- 不自行解离从而恢复成正常AChE,永久性降低AChE效能
- 常见毒物:有机磷酸酯类(organophosphate),如农业和环境卫生杀虫剂DDVP、dipterex等
- 毒理效应分析(围绕AChE被抑制,ACh过量激动N,M受体)
- 胃肠道摄入有机磷:胃部平滑肌过度激动,造成过度收缩引起痉挛绞痛,并有恶心呕吐腹泻
- 皮肤吸收有机磷:累及皮肤部位及附近区域大量出汗,肌束颤动
- 吸入或眼睛接触毒物:
- 针尖样瞳孔(M受体激动造成瞳孔缩小),严重近视
- 腺体分泌增加,支气管腺体、胃腺、唾液腺,出现流涎症状
- 呼吸道平滑肌过度收缩呼吸道收,加之呼吸道腺体分泌过多堵塞气管,引起呼吸困难
- 对中枢神经系统的影响:
- 先兴奋后抑制,先出现易激惹、惊厥等症状;随后迅速转变抑制效果,造成意识模糊、共济失调、昏迷、中枢性呼吸麻痹、中枢性血压下降
- M受体过度兴奋还会引起心脏活动下降和血管舒张。造成循环衰竭
- 作用于N受体的毒理作用(先兴奋后抑制)
- N-N受体:自主神经节先兴奋后抑制(激动效果过强,引起去极化抑制),症状有血压下降、心率减慢、大小便失禁
- N-M受体:先受体过度兴奋造成骨骼肌肌束震颤,抽搐痉挛;随后造成神经肌肉接头的突触后膜(骨骼肌细胞膜)的去极化阻滞,后期表现为肌无力和肌肉麻痹。进而引起呼吸肌麻痹 造成呼吸衰竭
- 毒理效应分析(围绕AChE被抑制,ACh过量激动N,M受体)
- 总体药效分析
- 实用意义
胆碱酯酶复活剂(解磷定、氯磷定)
-
机制
- 有机磷农药类物质(难逆性胆碱酯酶抑制剂),会和AChE形成牢固的共价键,从而导致AChE失去水解ACh的能力,从而ACh大量蓄积,引发胆碱能危象。所以使用胆碱酯酶复活药,使得AChE的功能复活,重新水解ACh。还应该使用M受体阻断剂(如阿托品),来降低胆碱能危象
-
肟类:与磷的亲和力更强(共价键)解脱酶使复活并直接结合游离有机磷(原理)
-
代表药物
- 碘解磷定:不稳定释碘,临用临配,碘刺激须静脉注射(少量多次,联合使用)
- 药动:肝代谢肾排出 < 1h 需重复给药
- 药理:拟胆碱药与体内胆碱酯酶结合,形成磷酰化酶而使之失去水解乙酰胆碱的作用,因而体内发生乙酰胆碱的蓄积。碘解磷定等解毒药在体内能与磷酰化胆碱酯酶中的磷酰基结合,而将其中胆碱酯酶游离,恢复其水解乙酰胆碱的活性
- 临床应用:作为抗胆碱药,部分杀虫剂(有机磷)中毒的解毒剂
- 疗效:与有机磷化学构型和中毒时间长短有关系(磷酰化胆碱酯酶“老化”前使用效果好),对肌肉颤动消除快,M症状慢需要阿托品(M受体非特异性拮抗药)足量及时对症,两者效果协同
- 中毒后,应该尽早使用药物,否则会引起AChE发生永久性的老化,无法恢复功能。
- 药物作用不是即刻生效,最开始表现为ACh的含量不再增加,随后缓慢下降。
- 临床实践中,药物效果并非完整,常常仅有中枢神经层面效应和N受体激动效应的缓解,一般还会搭配M受体阻断剂(阿托品)联用
- 不良反应:过量也会抑制AChE,加剧毒性。含碘使得咽痛腮腺肿大
- 氯解磷定:较碘解磷定优,可肌肉注射
- 碘解磷定:不稳定释碘,临用临配,碘刺激须静脉注射(少量多次,联合使用)
抗胆碱药
- 胆碱受体阻断药
- 基本概念和逻辑
- 理解“抗胆碱药”
抗胆碱药其实就是阻断突触后膜上ACh的M受体或者N-M或N-N受体的药物,其中,N-M受体是在骨骼肌接头处,不存在交换神经元的问题。N-N受体是在交感神经或副交感神经换元处的神经节上的,所以阻断了N-N受体的本质就是“阻断了神经节”,故如此命名。 - 理解“节后抗胆碱药”
由于M受体基本都位于有副交感支配的效应器上,即副交感节后纤维轴突和效应器之间形成突触的突触后膜上。不难理解如果阻断M受体,就会切断副交感神经节后纤维和效应器的联系。故如此命名。
- 理解“抗胆碱药”
- 神经节阻断药(N-N受体)
- 骨骼肌松弛药(N-M受体)
- 节后抗胆碱药(M受体)
- 作用效果
- 代表药物
- 阿托品以及阿托品类似药物(对全身M受体均有效果)
- 更具有选择性的M胆碱受体阻断药
- 阿托品(atropine)药理(包括类似物东莨菪碱————中枢抑制透皮吸收、山莨菪碱————不能通过bbb)
- 药动学
- 口服及粘膜均易吸收,全身分布,,维持效应4h(眼72h,房水循环慢),50%原型代谢,肝代谢,可以透过BBB及胎盘
- 作用部位:腺体、眼、内脏平滑肌、心肌、中枢神经系统
- 药效原理
- 对全身各处的M受体都有广泛的阻断作用,导致不同的组织器官的M受体激动度被抑制,从而引发副交感张力下降的效果。
- 药物效果
- 药动学
- 基本概念和逻辑
1.腺体抑制分泌
| 效果 | 应用 | ADR |
|---|---|---|
| 唾液 | 流涎症(ptyalism) | 口干 |
| 汗液 | 盗汗症(hyperhidrosis) | 皮肤干燥体温升高 |
| 支气管腺 | 全麻前给药 | 痰稠 |
| 胃酸分泌不敏感 | 哌仑西平(选择性阻断M1) | 实际组胺对胃酸分泌影响更大 |
(效果从高到低)
- Q1:为什么有“术前阿托品,术后新斯的明”的用药口诀?
手术前,预防性使用阿托品,避免患者产生吸入性肺炎。因为全麻病人不再具有咳嗽能力,而麻醉药物会刺激呼吸道造成患者呼吸道分泌物增加,可能出现分泌物堵塞气管的问题。所以使用阿托品阻断M受体减少支气管分泌物。手术后使用新斯的明,可以抑制胆碱酯酶,使得ACh的效应增强,可以加快消化系统和泌尿系统活动能力,便于术后排便排气
2.内脏平滑肌解除痉挛
| 效果 | 应用 | ADR |
|---|---|---|
| 胃肠道 | 胃肠绞痛(Gastrointestinal colic) | 腹胀、便秘 |
| 膀胱 | 遗尿症、尿频、尿急 | 小便困难 |
| 输尿道、胆道 | 肾、胆绞痛(加镇痛药如吗啡,效果理想) | |
| 支气管不敏感 | 异丙托溴铵(不增加痰稠) | |
| 膀胱M3受体选择性阻滞剂——索利那新 |
- Q2:为什么再治疗其他疾病而使用阿托品时候,一般饭时伴随水和纤维素食品服用?
由于阿托品会抑制M受体,造成胃肠道平滑肌活动减弱,使得胃肠道蠕动能力下降内容物堆积。因此通过食用纤维素食品和水来刺激胃肠道蠕动,以此来对冲不良反应
3.眼扩散瞳孔
| 效果 | 应用 | ADR |
|---|---|---|
| 扩瞳 | 虹膜睫状体炎 | 羞明 |
| 眼内压增高 | - | 青光眼禁用 |
| 调节麻痹 | 验光配镜,排除假性近视 | 视力模糊 |
| 抑制对光反射 | 眼底检查.恢复慢 | 托吡卡胺<后马托品<阿托品 |
眼科中现在较少使用阿托品,因为阿托品在眼中的作用时间过长(由于房水更新的时间过长,所以长期生效)
Q3:为什么使用阿托品类似的M受体阻断剂药物前,必须排除青光眼病史?
阿托品会阻断眼中的M受体引起睫状肌收缩瞳孔散大,这样会造成睫状体缩小,挤压前房角,使得前房角静脉窦回流房水的能力变弱,进而造成眼压升高,加剧对于眼底视觉神经的压迫,可能加重青光眼造成失明
4.心血管加快心率扩张血管
| 效果 | 应用 | ADR |
|---|---|---|
| 加快(恢复)心率,心脏活动强度上升(取决于基线) | 缓慢性心律失常、房室传导阻滞、窦性停搏 | 心悸 |
| 解除微血管痉挛和过度收缩(和阻断M受体无关,可能是代偿性散热) | 抗休克,改善循环障碍,避免重要气管缺血 | 面潮红 |
生理基础:M受体激动对心血管系统的影响————产生四负效应,降低心脏活动强度
药理效应:使用阿托品会加强心脏活动强度,心率、心肌收缩力全部上升。但是对于血管的影响不一样,在阿托品的使用下血管反而会舒张
阿托品化:血管会扩张,表现为皮肤的潮红和温热,瞳孔散大,口干皮肤干,心率加快。在对抗有机磷中毒时,使用阿托品剂量达到阿托品化足够用于解毒,为一般常用剂量的10倍以上
Q4:为什么休克伴有高热或心率过快的患者不可使用阿托品?
阿托品抑制M受体,使得汗腺分泌减少,患者的散热功能降低,可能引起极度高热,另外还会加快心率,可能进一步造成心动过速,引起更严重后果
5.CNS兴奋
| 效果 | 应用 | ADR |
|---|---|---|
| 兴奋呼吸 | 有机磷中毒抢救 | 烦躁、幻觉、惊厥 |
阿托品偏酸性,酸性条件下阿托品治疗效果受限
- 改进药物:戊乙奎醚,不影响心率,t1/2长达10h
- 阿托品不同剂量效果
- 治疗量:表现为中枢兴奋作用(可能是抑制外周M受体造成的代偿效果)
- 过量:表现为中枢神经兴奋性中毒症状,如焦躁、幻觉、共济失调、抽搐惊厥等
- 在过量:转而出现中枢神经系统的抑制,如昏迷、呼吸麻痹、循环和呼吸衰竭
- 解毒方法:毒扁豆碱对抗
- Q5:为什么阿托品过量中毒后应该使用毒扁豆碱解毒?
阿托品中毒后,大量M型受体被阻断,使用毒扁豆碱或者新斯的明可以抑制胆碱酯酶,使得ACh在突触间隙的含量升高,增强ACh的效能,间接对冲大量M受体被抑制的毒性。
胆碱受体阻断药(神经节阻断药)
-
作用效果的整体理解
- 特异性地阻断了N-N受体,就会导致交感神经节后纤维的张力和副交感神经节后纤维的张力都同时显著下降。剩下效果取决于以交感神经支配占主导还是副交感神经支配占主导。
- 对于心血管系统,交感神经支配占主导地位
- 血压下降
- 对于胃肠道、膀胱、腺体、眼睛等处,副交感神经支配占主导地位
- 消化道活动加强,引发腹痛、腹泻等;瞳孔缩小;唾液、胃液等分泌增加等
-
作用面广、副作用多、强度难控制现在不用
-
美加明、咪噻吩(阿方那特)
胆碱受体阻断药(骨骼肌松弛药)
N-M受体的本质是钠离子通道,有两种选择来阻断神经肌肉接头。常用于全麻辅助药
-
去极化型————强制打开钠离子通道
- 琥珀胆碱
- 生理原理:兴奋M受体,造成Na离子通道长期开放造成钠离子抑制内流,引发去极化阻滞,不能产生动作电位

- 体内代谢通路高效明确,会很快被胆碱酯酶水解,所以需要静脉滴注
- 作用特点:在肌肉麻痹之前,会有一段时间肌肉细胞兴奋性加强,产生短暂的肌束颤动
- 临床应用:侵入性操作的肌肉松弛,辅助麻醉药物
- 生理原理:兴奋M受体,造成Na离子通道长期开放造成钠离子抑制内流,引发去极化阻滞,不能产生动作电位
- 琥珀胆碱
-
非去极化型(竞争型)————与ACh竞争受体
- 筒箭毒碱(苄基异喹啉类(benzylisoquinolines))

- 阿曲库胺(氨基类固醇类(aminosteroid compounds))
- 相比筒箭毒碱:起效更快(1min);持续时间短(15min);不蓄积非酶降解,尿胆排泄(肝肾功能不全者适用);半衰期短(20min)
- 能够激动N-M受体的药物可以用来解毒本类药物,如新斯的明
- 作用特点:它一用药就是降低肌肉收缩,从而起到肌松效果的,不会有暂时肌肉震颤
- 同类药物:维库溴铵、泮库溴铵
- 筒箭毒碱(苄基异喹啉类(benzylisoquinolines))
-
肌肉松弛药的“多靶点作用”
-
琥珀胆碱:兴奋 M
-
筒箭毒碱:阻断 释放His
琥珀胆碱+筒箭毒碱联用效果?
会相互拮抗效果变差,原因是:非去极化阻断药物(筒箭毒碱)通过阻断ACh受体起作用,而去极化阻断药(琥珀胆碱)是ACh受体激动药
-
药物对肾上腺素能神经元信号传递的作用
肾上腺素受体激动药
去甲肾上腺素能神经(noradrenergic nerve):除了骨骼肌血管处和汗腺处以外的所有交感神经 节后纤维,都为肾上腺素能神经,以NE作为自己的神经递质。
- 拟交感胺药
- 分类
- 按结构:儿茶酚胺类和非儿茶酚胺类
- 按对受体选择性
- α受体激动药
- β受体激动药
- α和β受体激动药
- 三种肾上腺素的比较
- 构效关系
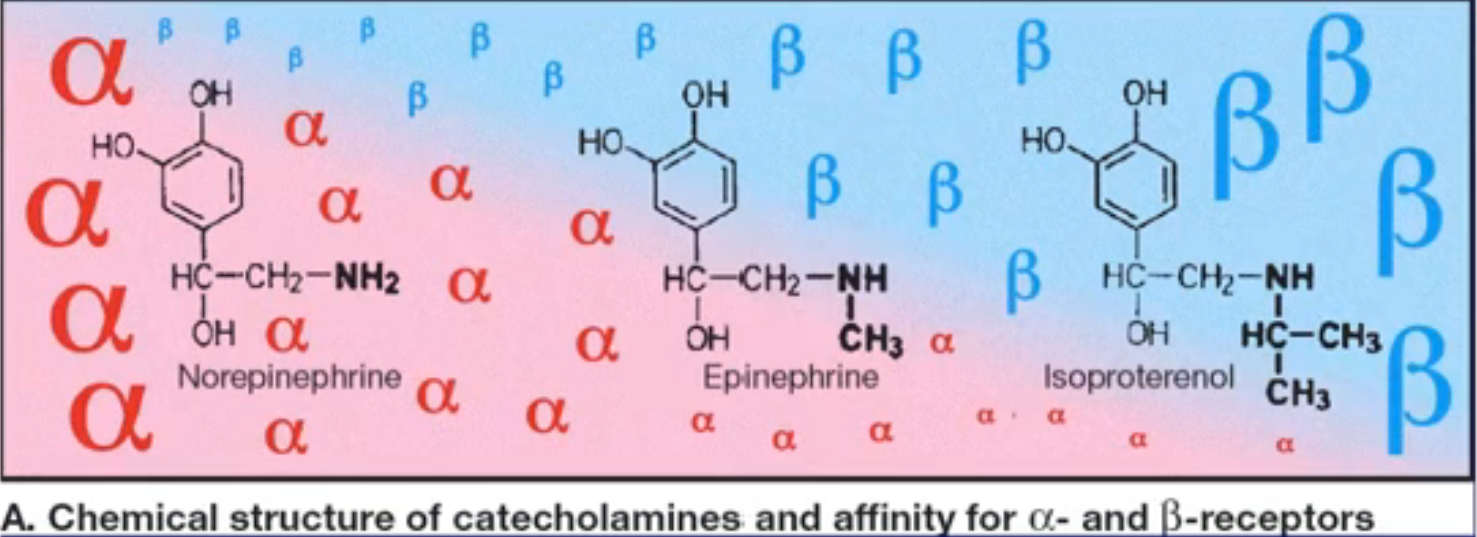
- 比较
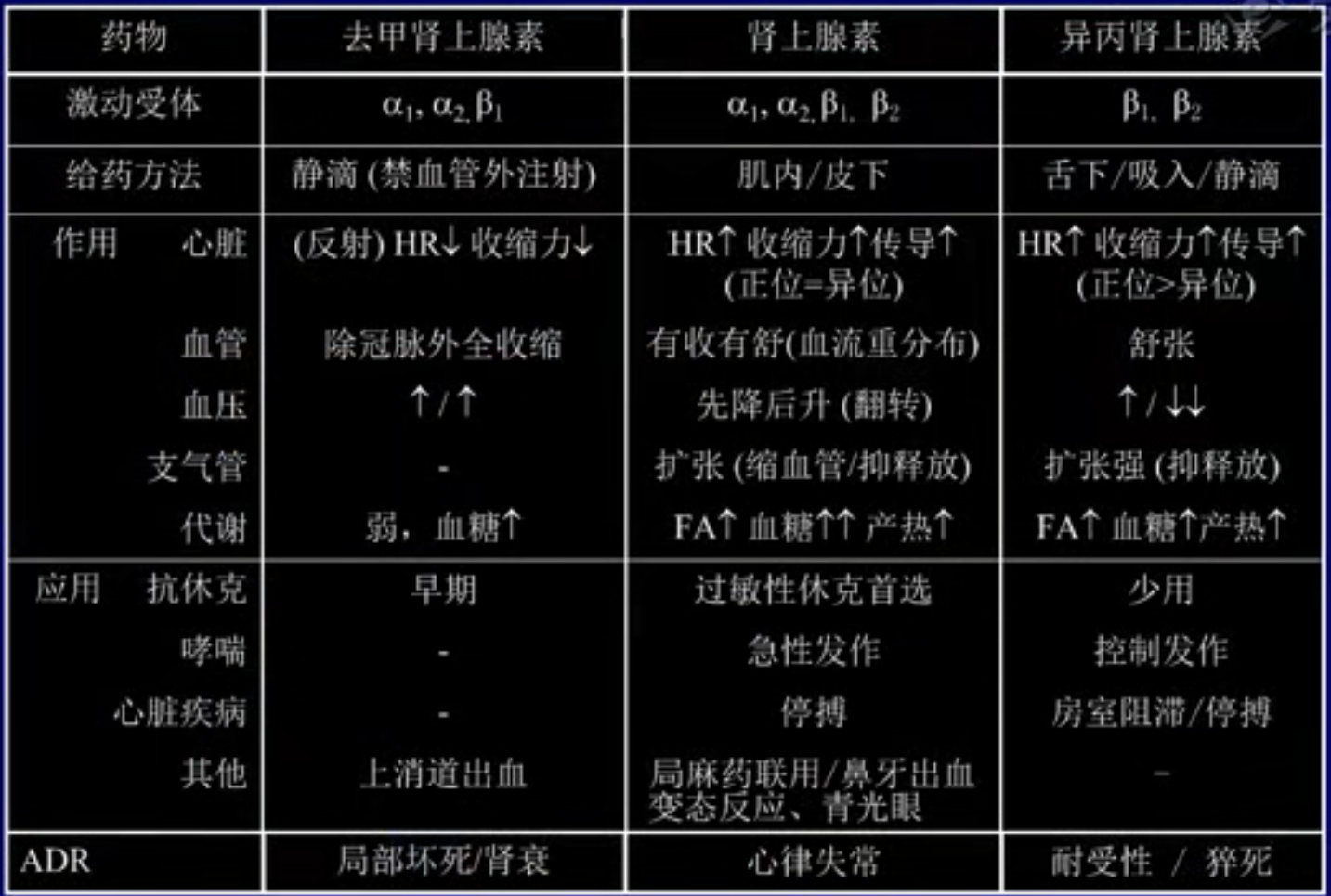
- 三肾激素共同的药理作用是:扩张冠脉血管、升高收缩压
- 肾上腺素加快心率是离体作用,在体是降低心率(收缩压升高,造成压力感受器兴奋,心率下降平衡血压
- 肾上腺素翻转:加入肾上腺素的效果与常见效果相反, 如先使用α受体抑制剂,再使用肾上腺素,降压效果会更加明显,而不是升高血压
- 生理基础:肾上腺素对α和β受体均有作用,比如对于血管来说,α受体兴奋起到收缩血管作用,β受体兴奋起舒张血管的作用。如果抑制α受体,等于解除了对β受体的对抗,而表现出更明显的降压
因此氯丙嗪过量中毒不能使用肾上腺素来升血压
该特性可以用于检查肾上腺髓质肿瘤:使用很小剂量的α受体抑制剂,检测是否有非常强烈的血压降低症状
- 构效关系
- 常见药物
- 间羟胺(阿拉明,可拉明)
- 直接作用为兴奋α受体,间接促进释放NA(细胞内递质有限,所以会快速出现耐药,受体表达量也下降)
- 早期休克可以取代NA
- 给药方式可以肌肉注射,还有静脉推注
- 多巴胺
- 作用 激动D受体(肾、肠系膜、冠状血管)及α、β1受体
1. 心血管 小剂量D受体扩大血管;大剂量α受体收缩血管;β1受体使得心率增加
2. D1受体扩张肾脏血管 肾小球滤过率增加 防止肾衰 排钠利尿;大剂量刺激α缩血管 - 临床:用于抗休克心衰急性肾衰
- 不良反应:恶心呕吐 心律失常
- 作用 激动D受体(肾、肠系膜、冠状血管)及α、β1受体
- 麻黄碱
- 口服皮下肌注均易吸收,能透过脑血屏障,作用持久
- 作用:激动αβ受体促进NA释放,作用弱而持久(不容易被酶破坏),容易产生快速耐受
- 药理效果
1. 心血管 心率上升,皮肤粘膜内脏血管收缩、骨骼肌血管扩张,血压上升。能够持续3-6h,不会产生继发性下跌
2. 支气管 松弛平滑肌,强度弱起效慢而持续久
3. 中枢神经 强兴奋皮质和皮质下中枢,过度兴奋可能造成失眠,弱兴奋呼吸循环中枢 - 临床:治疗鼻塞、缓解支气管哮喘、低血压等
- 不良反应:剂量不宜过多可能造成耐受,还可造成失眠头痛心悸
- 多巴酚丁胺 Dobutamine(β1受体选择性激动药)
- 不可口服,需要持续静脉滴注半衰期只有2min
- 药理效果:作为强效正心药物治疗急性心肌梗死、心衰、心肌收缩力减弱。
- 沙丁胺醇 Salbutamol、特布他林(β2受体激动药)
- 药理效果:作为平喘剂治疗哮喘,用于松弛支气管平滑肌,而不引起心脏作用。
- 间羟胺(阿拉明,可拉明)
- 分类
肾上腺素受体阻断剂
-
α受体阻断药
- 合用AD可以使升压转为降压,造成肾上腺素作用的翻转,对NA、异丙肾上腺素无影响
- 常见药物
- 具有受体选择性
- 哌唑嗪(α1阻断药) 抗高血压药物(不加快心率)
- 育亨宾(α2阻断药)
- 无选择性
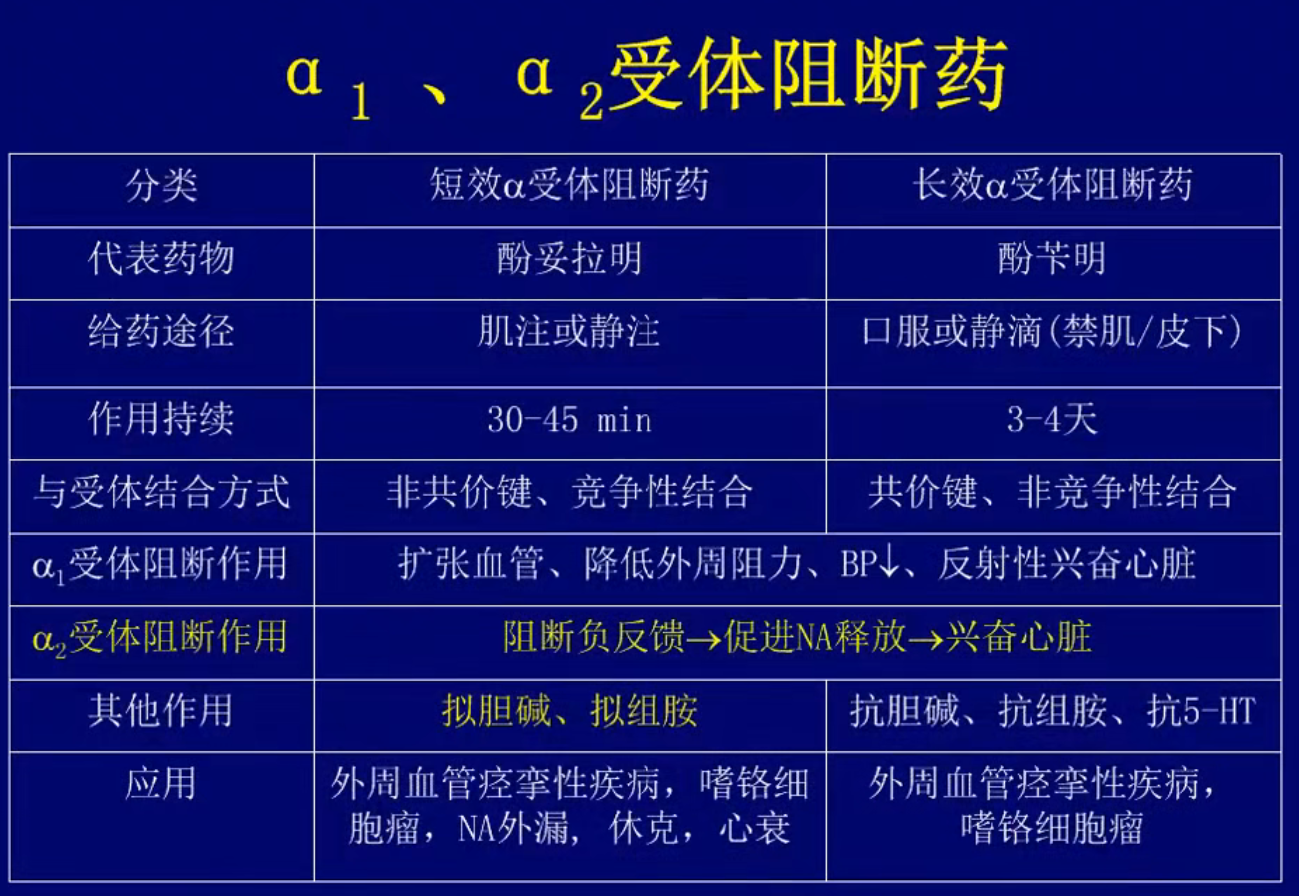
α受体阻断药不适用于高血压的主要原因:该药物只无选择性地阻断了α受体,但是去甲肾上腺素的β受体没有阻断。这就造成了,血压下降的同时,心率有显著性提升
如果选择性阻断α1受体,这样会保留α2受体的反馈作用,从而减少了提升心率的副作用,更适合作为降压药物 - 具有受体选择性
- 案例分析
酚妥拉明和酚芐明都有恶心呕吐腹痛的不良反应,前者口服/注射均发生,而后者只见于口服给药时,试分析其原因:
酚苄明仅口服时有不良反应,说明这是局部的作用,该药物有强烈的胃肠道刺激性,如果直接进入组织也会引起强烈的组织疼痛;而酚妥拉明是一个全身作用,具有拟胆碱拟组胺的效果,会兴奋胃肠道平滑肌,可能会引起上述不良反应
在缓解的时候,酚苄明由于具有抗胆碱抗组胺的效果,因此如果使用阿托品来缓解不良反应效果是不好的,但是可以用于缓解酚妥拉明的不良反应 -
β受体阻断药
- β受体阻断作用
- 心脏β1受体:心率/心传导/心力/心输出/心耗氧下降/有效不应期上升
- 支气管β2受体:收缩平滑肌诱发加重哮喘
- 代谢β2:低血糖无症状
- 肾素β1:抑制球旁细胞肾素释放
肾素-血管紧张素-醛固酮系统为体内肾脏所产生的一种升压调节体系,引起血管平滑肌收缩及水、钠潴留,产生升压作用,是维持高血压的主要机制
作为药物,对心脑肾的阻断是产生治疗效果的重要基础,而对代谢支气管的干扰是引起不良反应的基础
- 内在拟交感活性 可减少支气管收缩、心衰、传导阻滞————说明对β受体的阻断作用并不完全
- 膜稳定 抑制细胞膜离子通透性(仅过量有效)
- 主要临床应用(降低高血压,需长期服用)
- 短期使用:心脏抑制,血管收缩,血压不变
- 长期用药:心脏抑制血管舒张,外周阻力下降,收缩压舒张压均下降
- 代表药物
- 普萘洛尔(非选择性的β1β2阻断)
- 药动学:口服易吸收,有明显首过效应,存在较大个体代谢差异(酶活性个体差异)
- 临床应用 心绞痛、心律失常、高血压、甲亢、心衰、青光眼等
- 不良反应
- 恶心乏力失眠多梦(干扰5-羟色胺)
- 恶化心功能不全,诱发哮喘
- 骤停诱发高血压心律失常心绞痛,应该逐渐减少剂量————症状反跳
- 噻吗诺尔 最强效用于青光眼降低眼压
- 美托洛尔(选择性β1受体阻断剂,仅亲和力高于β2)
- 心脏选择性强 持久
- 哮喘糖尿病患者慎用
- 克仑特罗(Clenbuterol): 强效选择性β2-受体激动剂,过量时也可使得β1受体兴奋。瘦肉精的成分,临床上常用于解除支气管平滑肌痉挛(抗哮喘),长时快速支气管扩张剂,实现长时平喘。不良反应:心动过速
- 克伦特罗临床停用原因:舒张颈动脉(β2)EC50=6.3nM,增加心率(β1)EC50=38nM,治疗指数(选择性)仅为β1(LC50)/β2(EC50)=6
- 沙丁胺醇(Salbutamol): 治疗指数=162,选择性更高更安全,目前临床仍然使用
- 普萘洛尔(非选择性的β1β2阻断)
- β受体阻断作用
总结
对于作用于肾上腺素能神经元信号传递的药物,应该结合其生理过程进行记忆和理解。
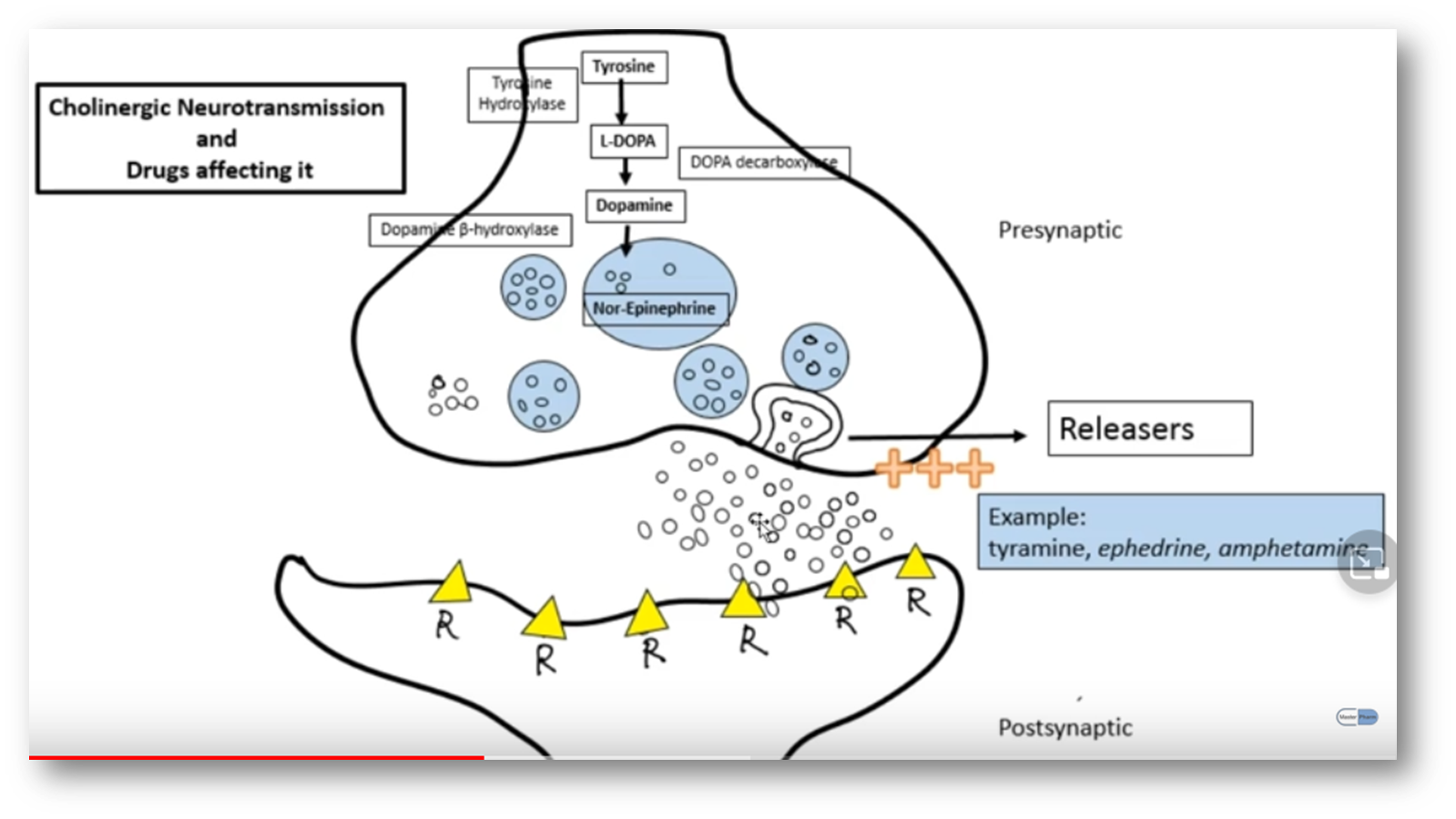
该图说明了去甲肾上腺素从合成到释放再到发挥作用最后被降解的全过程。结合我们所学的药物知识,这些药物都根据去甲肾上腺素的生命周期在不同时点发挥作用。比如利血平作用于节前细胞中NE运输入囊泡的转运载体蛋白,从而减少突触小泡中的NE数量,进而抑制神经活性;常见的激动剂拮抗剂都是定位于递质的释放以及突触后膜的受体产生作用;新斯的明作用胆碱酯酶抑制剂,抑制NE的降解步骤,从而增加在突触间隙NE的浓度;可卡因通过抑制突触前膜上的NE转运蛋白,抑制NE的重吸收来增加NE浓度,提高神经活性。
因此结合生理结构和过程,有助于增加对药物效果的深入认识。
抗高血压药理
心脏的电生理
心室肌电活动
-
心肌和其他兴奋性组织的区别
- 心肌在起搏点活动
- 动作电位长,且有平台期
- 平台期有内流,引发动作电位
-
心肌细胞动作电位阶段
- 静息电位
- 膜表面Na+K+泵构建胞内高钾、低钠环境
- 由K+外流,Na+内流形成电位差,即静息电位(-90mV)
- 0:快速去极化期(fast depolarization)
- 膜电位达到阈电位(-70mV)时,钠离子电压门控通道(INa,快钠通道)短暂开放,内向钠电流增大,形成一个正反馈的去极化,达到峰电位。随后钠离子电压门控通道关闭,直到下一次去极化
- 1:部分复极化期(partial repolarisation)
- Ito(K+外流)开放,形成瞬时外向电流
- 2:平台期(plateau)
- 平台期是心室肌与神经骨骼肌的最大区别
- 本质属于复极化过程,由Ik1、Ik、ICa-L完成
- 平台期形成的关键因素
- L型钙离子通道介导的Ca2+内流(CICR)
- Ik1非门控内向整流钾通道(长期开放)介导的k+外流减少
- Ik延迟整流钾通道,随时间缓慢增加外向K+电流。有利于进入复极化期
- 3:复极化期(repolarisation)
- Ik和Ik1促进K+外流,加速复极化
- 注意这里Ik1不再起内向整流减少K+外流,而是相反
- 4:起搏点(pacemaker)
- 本质上是恢复到静息状态的化学成分,将流入细胞质的Ca2+和Na+排出,而使得K+内流
- NaCa交换,Ca泵,Na泵发挥功能
- 80%以上Ca2+被肌质网回收,20%钙泵被排出体外
- 静息电位
-
0期的意义:区分快反应细胞 & 慢反应细胞
- 快:心室肌、心房肌、浦肯野纤维
- 0期由快钠通道介导Na+内流(INa)
- 去极化速度幅度大,电位差大局部电流大,电位传导快
- 慢:窦房结、房室结
- 0期由L型慢钙通道介导Ca2+内流(ICa-L)
- 去极化速度幅度小,电位差慢局部电流小,电位传导慢
- 快:心室肌、心房肌、浦肯野纤维
房室结的电位传导最慢,因此在临床上非常容易在房室结处出现传导阻滞,引起心律失常
-
4期的意义:区分工作细胞 & 自律细胞
- 工作细胞:能完成心室心房收缩的细胞————心室肌、心房肌
- 无4期自动去极化
- 只负责完成收缩功能
- 自律细胞:窦房结、浦肯野纤维…
- 有4期自动去极化
- 构成心脏的传导系统
- 工作细胞:能完成心室心房收缩的细胞————心室肌、心房肌
-
常见阻断剂
- Ik1:钡
- INa
- 河豚毒素(对神经细胞敏感性高)
- I类抗心率失常药(IA奎尼丁、IB利多卡因、IC普罗帕酮):降低0期去极化速度和幅度,降低传导速度,适用于室上性快速型心率失常
- Ito:氨基吡啶
- ICa-L:CCB(维拉帕米,不影响骨骼肌上的钙通道通过变构释放钙离子)
- Ik:III类抗心率失常药(胺碘酮)、四乙铵
-
平台期意义
- 增加了心室肌动作电位时程(APD)明显变长(从0期去极化开始到3期复极化完毕)
- 使得不应期明显变长(持续整个收缩期和舒张早期)
- 让心室肌几乎单收缩(利于心脏射血,避免强直收缩带来的僵硬)
窦房结P细胞电活动
-
区别心室肌
- 没有平台期(1、2期因为膜上不存在对应通道)
- 4期存在自动去极化,到达阈电位(-40mV)进入0期去极化
- 最大复极电位为-70mV,本质上和静息电位没有区别
为什么窦房结最大复极电位小于心室肌静息电位?(通透性,电化学驱动力)
- 因为膜上IK1通道数目更少
- 0期去极化速度幅度慢于心室肌————ICa-L(慢于INa)
- ICa-L大量开放的阈电位(-40mV)
- INa大量开放的阈电位(-70mV)
-
窦房结4期自动去极化
-
去极化速度最快(自律性最高)
-
自动去极化机制
- K+外流逐渐减少(主要机制)————IK激活程度逐渐减小(心室肌IK逐渐激活)
- Na+内流逐渐增多————If(慢钠通道,激活失活慢):复极化达到-60mV开始激活,超极化到-100mV充分激活
一般地,根据电压实验结果,超极化不会改变电导,去极化改变电导,因为会激活通道开放。
使用选择性窦房结If抑制剂(伊伐布雷定)————减慢心率,因为窦房结控制心跳
区别于β受体阻断剂美托洛尔,后者会造成四负效应,而前者没有这些副作用- Ca2+内流逐渐增多————ICa-T(T型快钙通道)
-
-
总结Ca2+通道
- L型慢钙通道
- 分布:占多数(肌膜/T管膜、心肌快反应细胞2期平台期、心肌慢反应细胞0期、消化道平滑肌0期)
- 阈电位:-40mV
- 阻断剂:CCB(维拉帕米)
- T型快钙通道
- 分布:窦房结4期自动去极化
- 阈电位:-50mV
- 阻断剂:Ni2+
- L型慢钙通道
-
三种心肌细胞小结
| 心室肌 | 窦房结 | 浦肯野纤维 | |
|---|---|---|---|
| 静息最大复极电位 | -90mV | -60mV | >-90mV |
| 0期去极化 | INa(快反应细胞) | ICa-L(慢反应细胞) | INa(快反应细胞) |
| 1期部分复极化 | Ito(k+外流) | 无 | Ito(k+外流)相较心室肌更明显 |
| 2期平台期 | ICa-L,IK,Ik1 | 无 | 通道更发达,比心室肌平台期更长 |
| 3期复极化 | Ik1 K+外流 | K+外流 | K+外流 |
| 4期 | 无自动去极化(工作细胞) | 自动去极化(ICa-T/If Ca2+,Na+内流增加,IK+ K+外流减少——主要效果)自律性最强 | 自动去极化最慢(If Na+内流增加持续引起超极化——主要效果,Ik+ K+外流减少) |
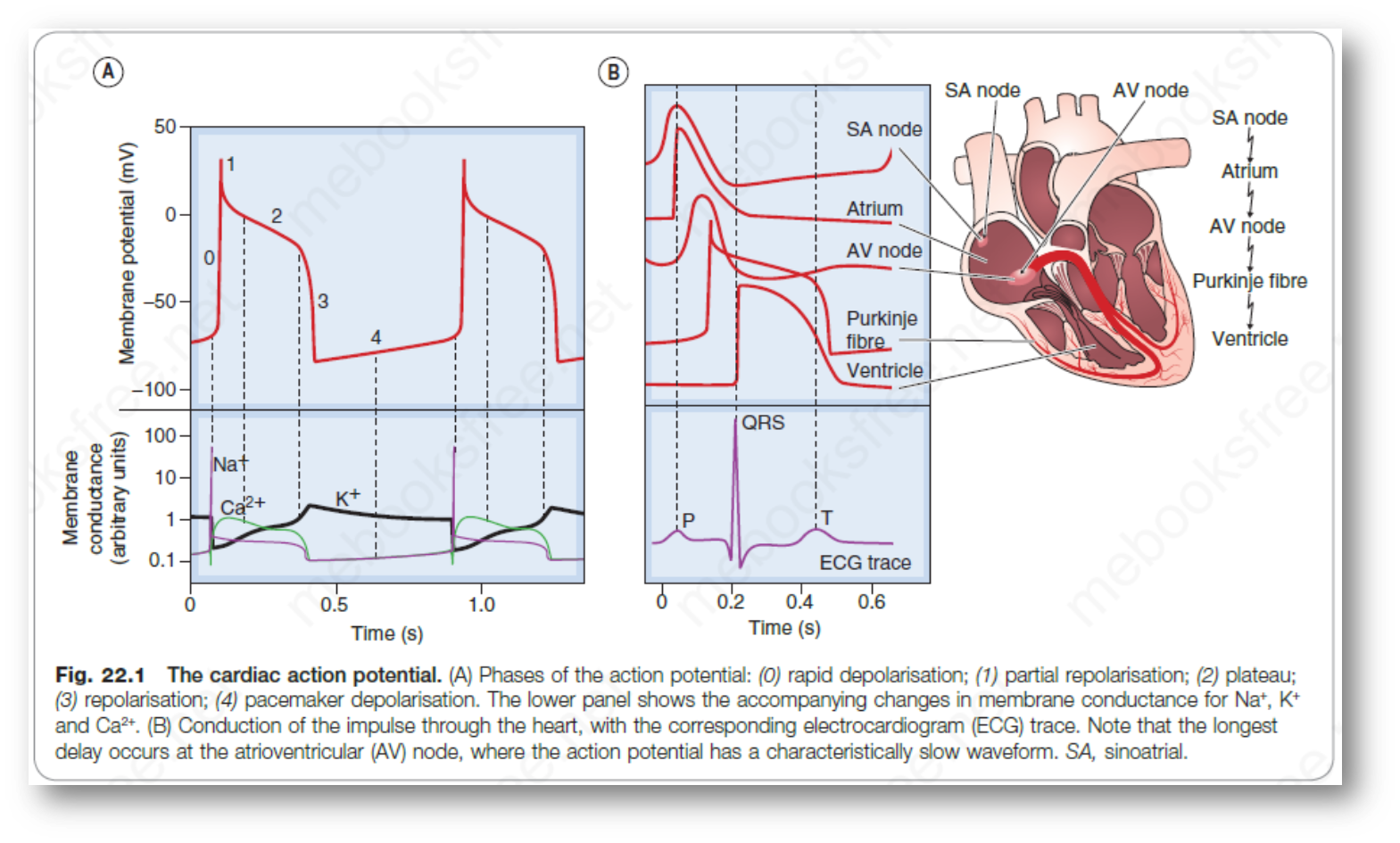
心肌的电生理特性
(1) 兴奋性
-
复习神经肌肉细胞的兴奋性
- 绝对不应期:由于电压门控钠通道在激活之后很快失活,必须复活才可再次被激活,恢复活性的时间产生不应期。该阶段任意刺激都不能激活钠通道。
- 相对不应期:细胞膜上的复活的钠通道数目远少于正常细胞,因此兴奋性更低
- 超常期:大部分钠通道活性恢复,由于膜电位与阈电位相近,使得超常期兴奋性高于正常
- 低常期:由于钠钾泵生电作用,使得膜电位和阈电位变远,兴奋性下降
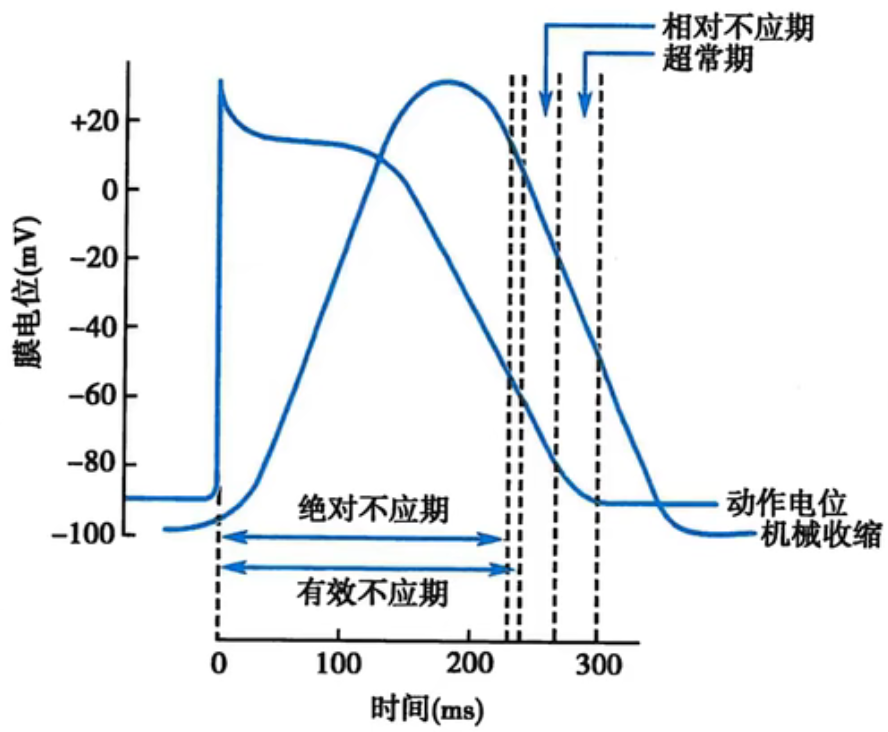
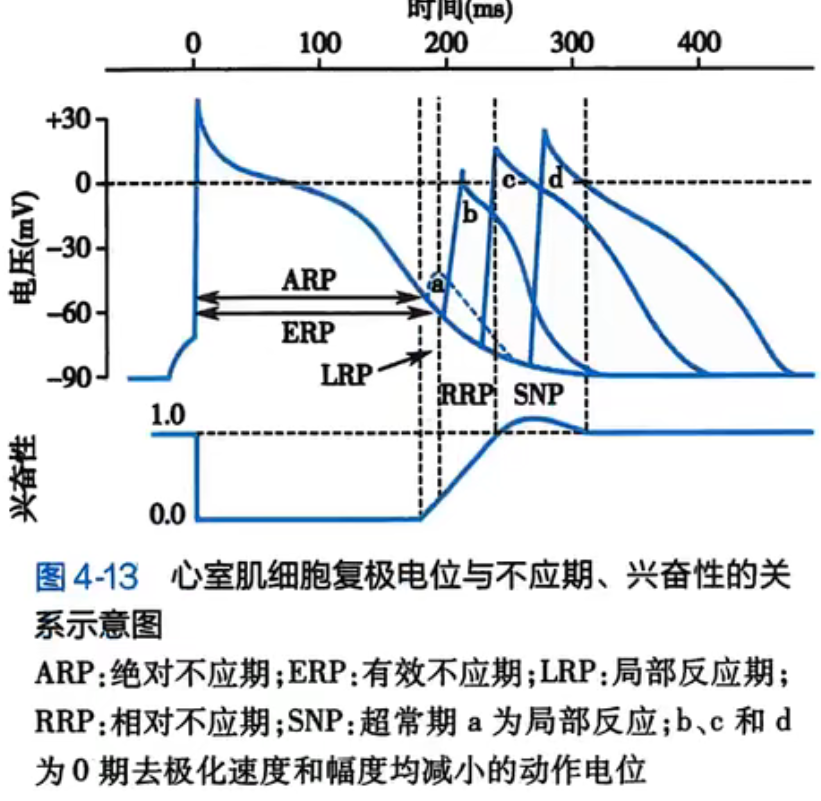
- 有效不应期(ERP)————给任何刺激都不会反应
- 绝对不应期(ARP)
- 局部反应期(LRP)————局部钠通道有相应但不能形成动作电位
- 相对不应期(RRP)————形成的动作电位弱峰电位低,对刺激不敏感
- 超常期(SNP)————峰电位仍然弱于静息状态(因为通道没有完全恢复活性)
- 无低常期
- 原因:心室肌的4期不存在正后电位,因为此时膜上同时有钙泵,钠钙交换泵和钠泵同时工作,前者向胞内增加正电荷,后两者和效应移除正电荷,两者效应几乎抵消,因此膜内外不存在正后电位。而神经骨骼肌细胞因为4期主要只有钠泵活动加强,所以会形成正后电位(膜电位轻度超极化)
-
分析兴奋性
- 膜电位和阈电位距离
- 距离远:兴奋性下降
- 距离近:兴奋性上升
- 距离过近:兴奋性下降————内外K+浓度差过小,胞膜快钠通道失活,引起去极化阻滞
- 膜电位和阈电位距离
-
动作电位时程(APD)和有效不应期(ERP)的关系
- IA奎尼丁:延长ERP > 延长APD -> 上升 -> ERP相对延长
- IB利多卡因:缩短ERP < 缩短APD -> 上升 ERP相对延长
因为两者都使得ERP相对延长,因为对ERP和APD的处理程度不同。有效不应期的增加可以使得更多动作电位都落在该时间区间内,所以可以降低心率。
(2) 传导性
- 传导系统构成:窦房结 -> 结间束 -> 房室结区 -> 房室束/希氏束 -> 左右束支 -> 浦肯野纤维
- 传导特点:
- 传导最快的是浦肯野纤维,保证各个房室肌细胞能几乎同时接收到动作电位
- 传导最慢的是房室结:存在房室延搁现象,意义在于便于心脏先进行回血在进行射血(物理层面理解)
- 分析比较传导性:
- 0期去极化的速度和幅度,越高电位差越大局部电流越强传导性越强
- 如果膜电位降低,钠平衡电位和静息电位差距下降,0期去极化速度和幅度下降,则引起传导性下降
- 如果细胞外Na+/Ca2+增加,即胞内外浓度差增加,ENa/ECa的平衡电位增大,引起去极化速度幅度变大,传导性增强
- 邻旁未兴奋细胞的兴奋性
- 生理结构
- 心肌细胞直径越大,电阻越小,传导速度越快:如浦肯野纤维细胞直径70um,传导速度最快;而房室结细胞直径3um,传导速度最慢
- 心肌细胞间的润盘(缝隙链接)越多,传导性越高
- 心肌细胞的分化程度越高,传导性越高
- 0期去极化的速度和幅度,越高电位差越大局部电流越强传导性越强
(3) 自律性
- 自律性大小:窦房结(100次/分,受到迷走神经的调节)> 房室结(50次/分)> 浦肯野纤维(25次/分)
- 由于窦房结自律性最高,所以称为正常起搏点,其他自律细胞为潜在起搏点
-
抢先占领:自律性更高的细胞快于下位细胞直接产生刺激,直接激活还没有达到阈电位的下位细胞
-
超速驱动压抑:自律细胞在受到更高频率的兴奋刺激的时候就会按照高频率信号产生动作电位。高频率刺激结束之后,下位细胞在恢复本身自律频率过程中需要一定的缓冲时间
-
举例:不能突然终止起搏器否则会出现心脏骤停
-
潜在起搏点何时表现自律性?
- 窦房结病变————逸搏
- 窦房结正常,潜在起搏点异常增高————早搏
- 有效不应期之后(相对不应期)
- 下一次窦房结兴奋传来之前
- 心室肌受到有效刺激
- 早搏之后心肌有较长的舒张期,即代偿间歇(窦房结兴奋恰好落在期前的ERP内)
-
分析自律性
- 4期自动去极化速度,如4期自动去极化中的内向电流增加,则去极化速度加快,自律性增强
- 最大复极减小,和阈电位举例变近,自律性增加
- 细胞外钙离子浓度增加,阈电位增加,最大复极电位距离变远,自律性下降
- 阈电位的影响因素:电压门控钠通道的密度状态和胞外钙离子浓度
(4) 收缩性
- 工作细胞的收缩特点
- “全或无”:润盘(同步化活动/功能合胞体)
- 单收缩:平台期 -> ERP长
- 依赖细胞外的钙离子(CICR)
- 分析收缩性
- 平台期钙离子内流的速度或量增加 -> 胞质钙离子增加 -> 活化横桥数目增加 -> 心肌收缩能力增加
心肌电生理小结:
通道小结: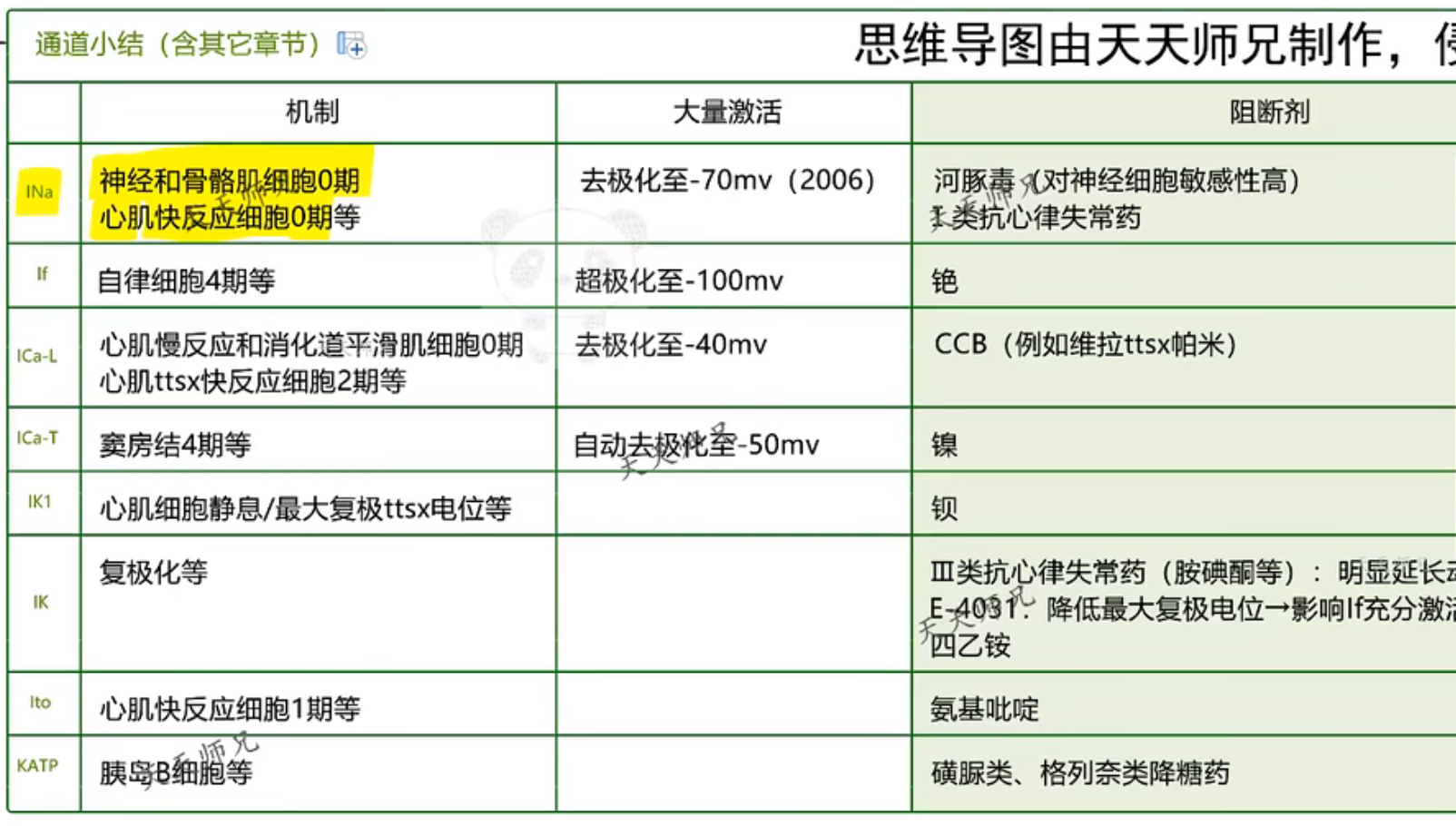
常见药物对心肌电生理的影响
- 儿茶酚胺(CA)———— 去甲肾上腺素NE|肾上腺素Adr
- 增强收缩力
- CA促进工作细胞 平台期ICa-L -> Ca2+内流增大 (CA激动β1-R -> G蛋白 -> AC -> cAMP增多 -> PKA -> 增加钙通道开放频率)
- 加快传导性
- CA促进慢反应细胞0期 ICa-L -> 0期 去极化速度和幅度增加
- 增快心率
- CA促进 窦房结 4期 ICa-T 和If -> 4期自动去极化速度增大 -> 自律性增大
- 增快舒张速度
- Ca使得钙泵对钙离子的亲和力增大 -> 回收钙离子加快
- 增强收缩力
- 乙酰胆碱(Ach)
- 减弱收缩力 -> 平台期缩短,平台期Ca2+内流下降
- Ach抑制工作细胞平台期ICa-L
- 促进Ik-Ach(主要是在心房肌)
- 减慢传导性
- Ach抑制慢反应细胞0期ICa-L -> 0期去极化速度和幅度下降
- 减慢心率 -> 4期自动去极化速度下降————自律性下降
- 促进Ik-Ach —> 最大复极电位超极化 -> 与TP距离变远
- 乙酰胆碱抑制窦房结4期ICa-T和If
- 减低兴奋性
- Ach促进Ik-Ach -> 膜电位与阈电位距离变远
- 减弱收缩力 -> 平台期缩短,平台期Ca2+内流下降
- 维拉帕米 ———— 阻断L型慢钙通道
- 减弱收缩力(负性肌力作用)
- 抑制工作细胞2期ICa-L -> 平台期钙离子内流减少
- 减慢传导性(负性传导作用)
- 抑制慢反应细胞0期ICa-L -> 0期去极化速度和幅度下降
- 减慢心率(负性频率作用)
- 维拉帕米(同E-4031)可以阻断IK -> 降低窦房结最大复极电位 -> 影响If激活
- 减弱收缩力(负性肌力作用)
高血压概述
基本概念
- 定义:在未使用降压药物的情况下,有三次诊室血压值均高于正常(缩压≥140mmHg或舒张压≥90mmHg),且这三次血压测量并不在同一天内进行,就可以诊断为高血压
- 分类:原发性、继发性高血压
生理机制
- 对于收缩压和舒张压的理解
- 收缩压:心脏的输出量和射血能力的高低————心率越快、心搏出量越大、心肌收缩力越强,则收缩压越大。
- 舒张压:外周血管阻力的大小————外周血管阻力越大(血管舒张程度大小)、血管顺应性越低(血管弹性越低),则舒张压越高
- 案例分析:老年人高血压的主要原因
- 一半老年人的心脏输出量没有异常增高,但是由于个体衰老,外周血管的顺应性(弹性)下降,并且存在部分血管狭窄。从而引起外周血管阻力增加,集中体现在舒张压升高, 但收缩压不升高。因此药物选择上一半选用舒张血管的药物,而不是降低心输出量。
- 对血压调节的生理系统
- RAAS系统:如果患者血容量下降到低于血管床容量的水平,则可以激动球旁细胞分泌更多的肾素从而激动RAAS系统,进而起到增加AngII和醛固酮的分泌量的效果。前者促进全身血管收缩,后者引起水钠潴留,增加血容量。
- 交感神经-肾上腺素系统:血压过低会激动交感神经,进而激动肾上腺髓质,促进分泌更多儿茶酚胺类物质,如肾上腺素,促进血管平滑肌收缩,从而提升血压
- 治疗思路:抑制患者过度激动的RAAS系统或者交感-肾上腺素系统激动程度
常见抗高血压药
总述
- 五大一线降压药————AABCD
- ACEI、ARB、β受体阻断剂、CCB、利尿剂(Diuretics,并且在降血压的时候主要选用其中的“噻嗪类利尿剂”,别的利尿剂一般不用,因为利尿强度不适合)
血管紧张素转化酶抑制药(angiotensin-converting enzyme inhibitor,ACEI)
抗高血压药物总结
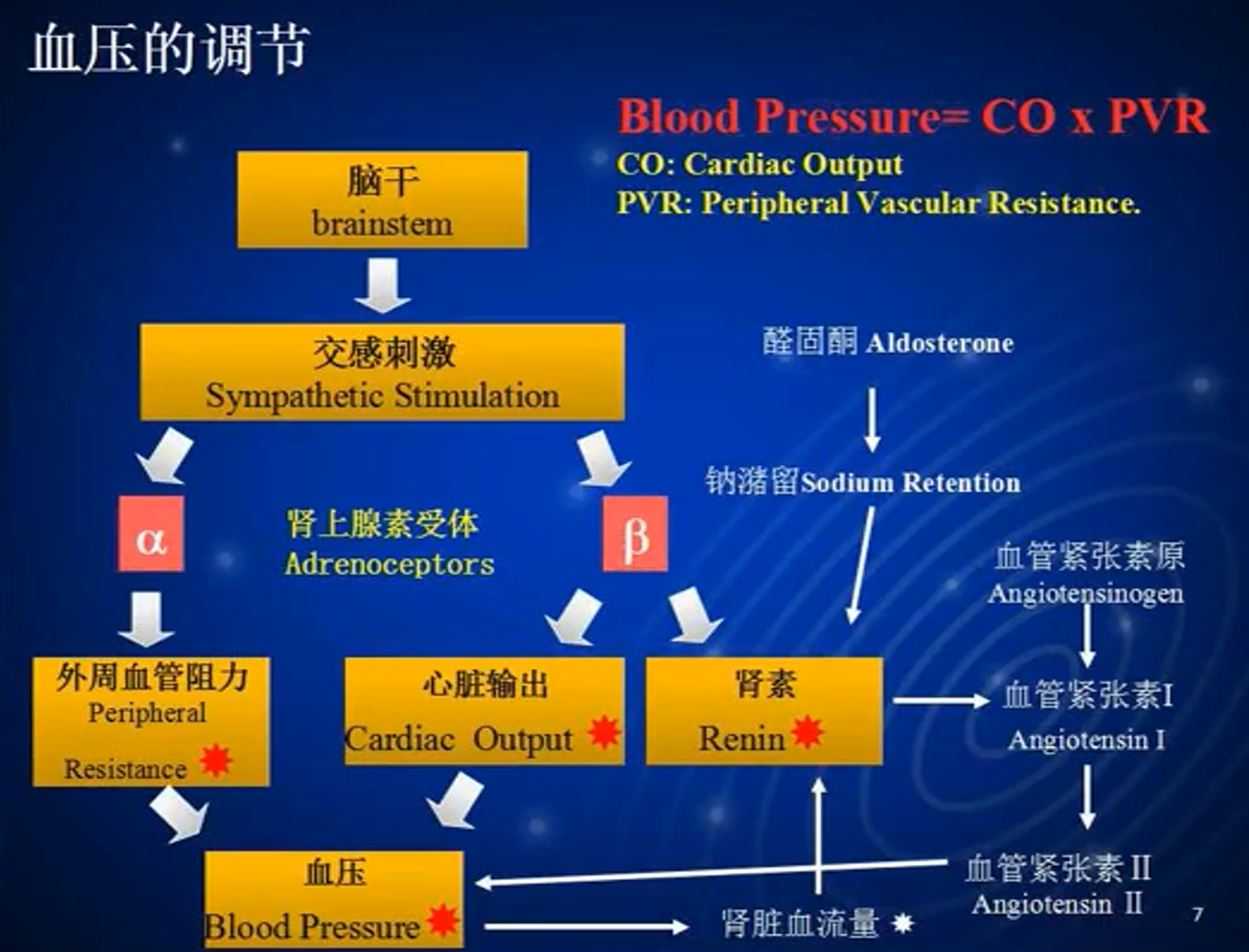
针对血压调节阶段的不同抗高血压药物
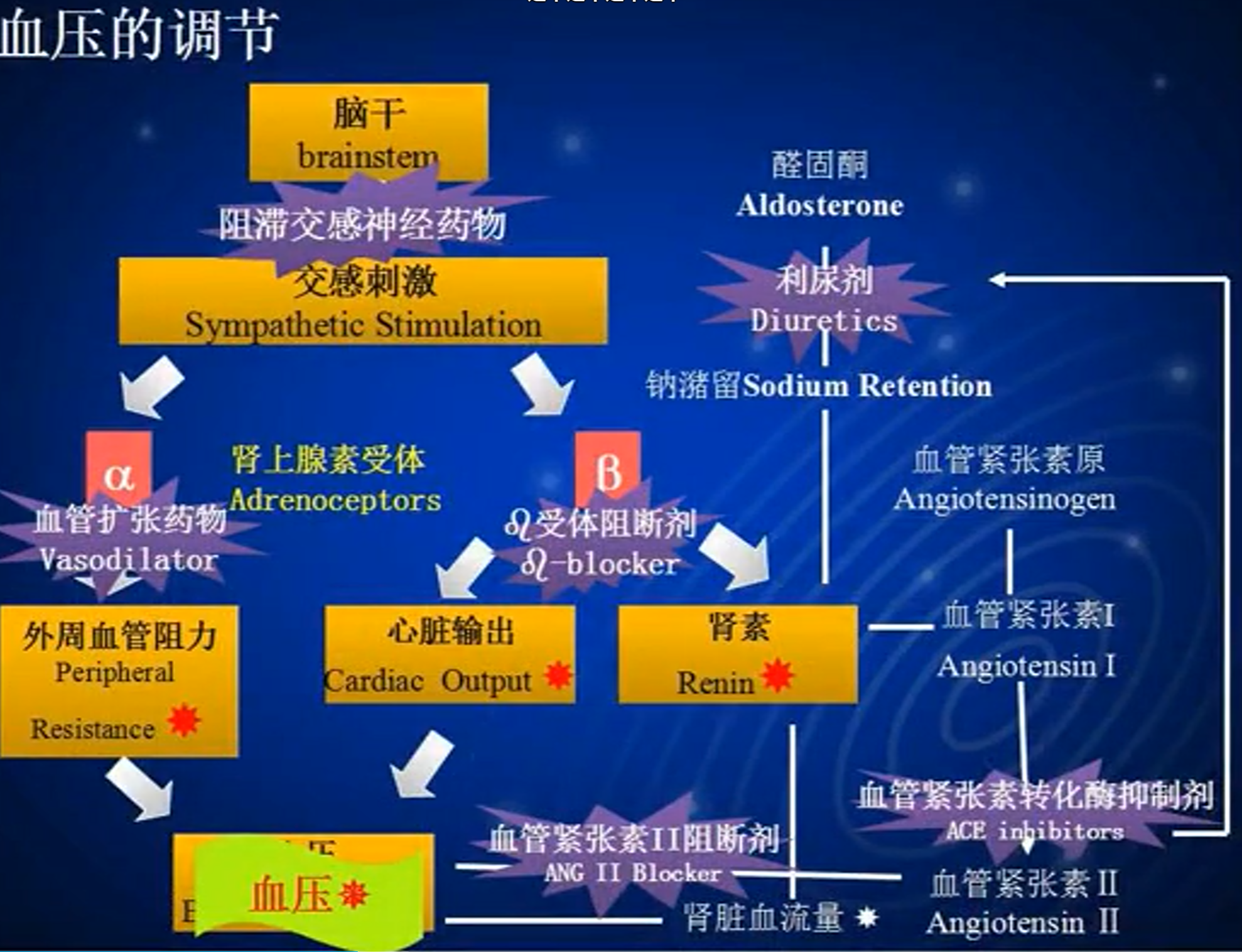
- 抗高血压药物的分类
- 肾素-血管紧张素系统抑制药
- 肾素-血管紧张素系统分子机制及作用靶点
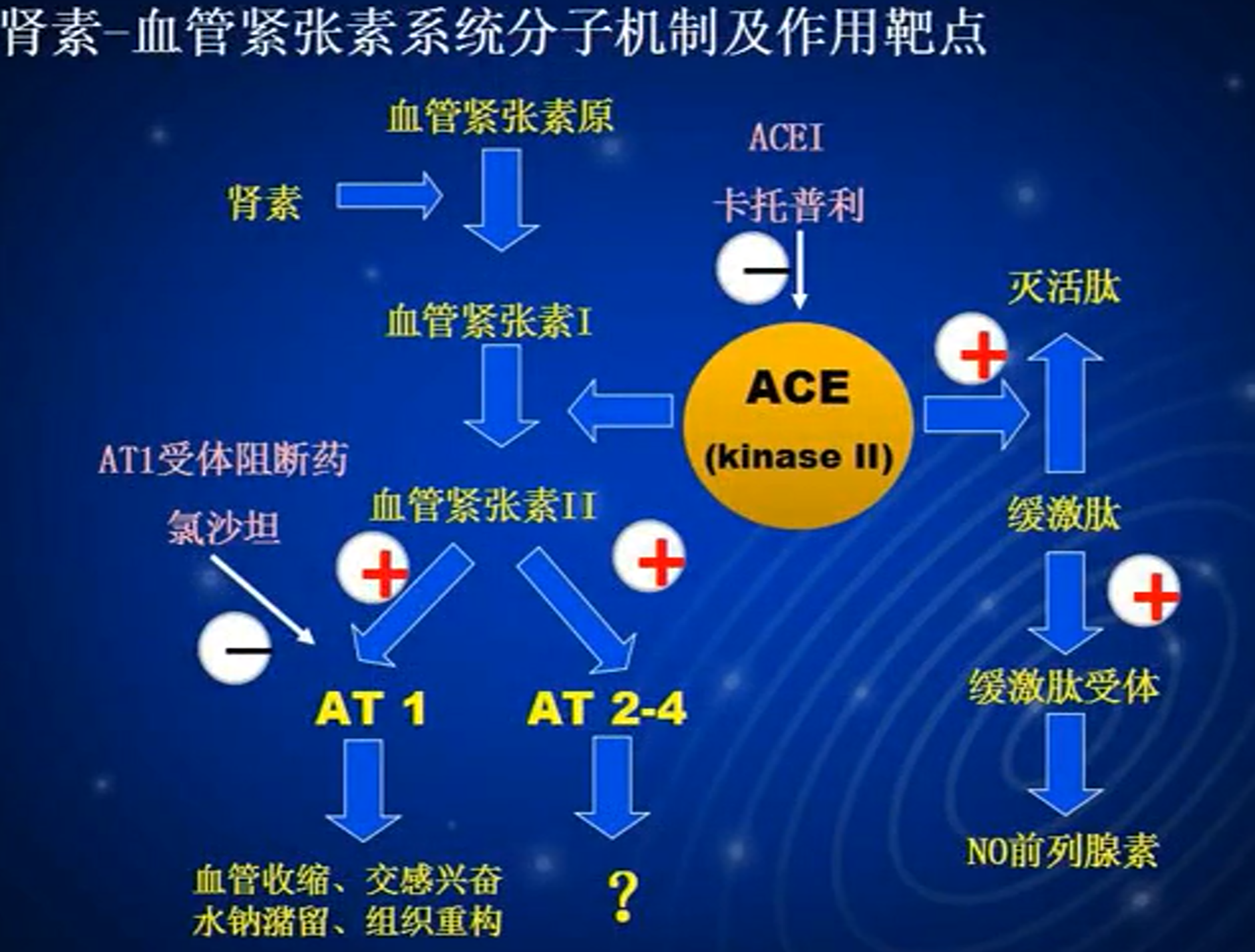
- 血管紧张素酶的抑制药物

- ACEI药理作用


- 作用机制

- 临床应用

- 不良反应

- 常用药物
- 氯沙坦 Losartan

- 依那普利 Enalapril

- 氯沙坦 Losartan
- 肾素-血管紧张素系统分子机制及作用靶点
- 通道阻滞药
- 作用机制
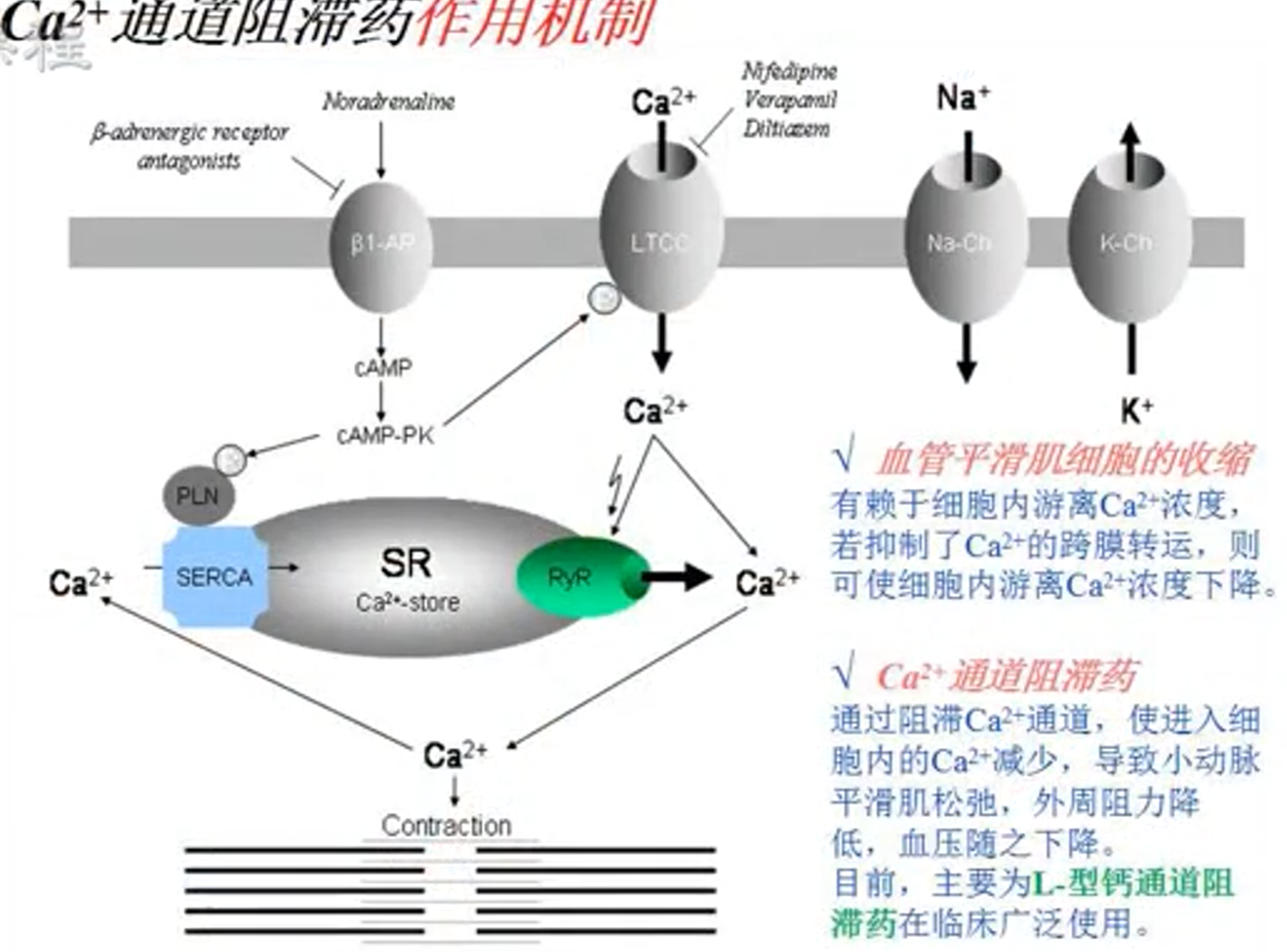
- 药理作用

- 临床应用和不良反应

- 常用药物
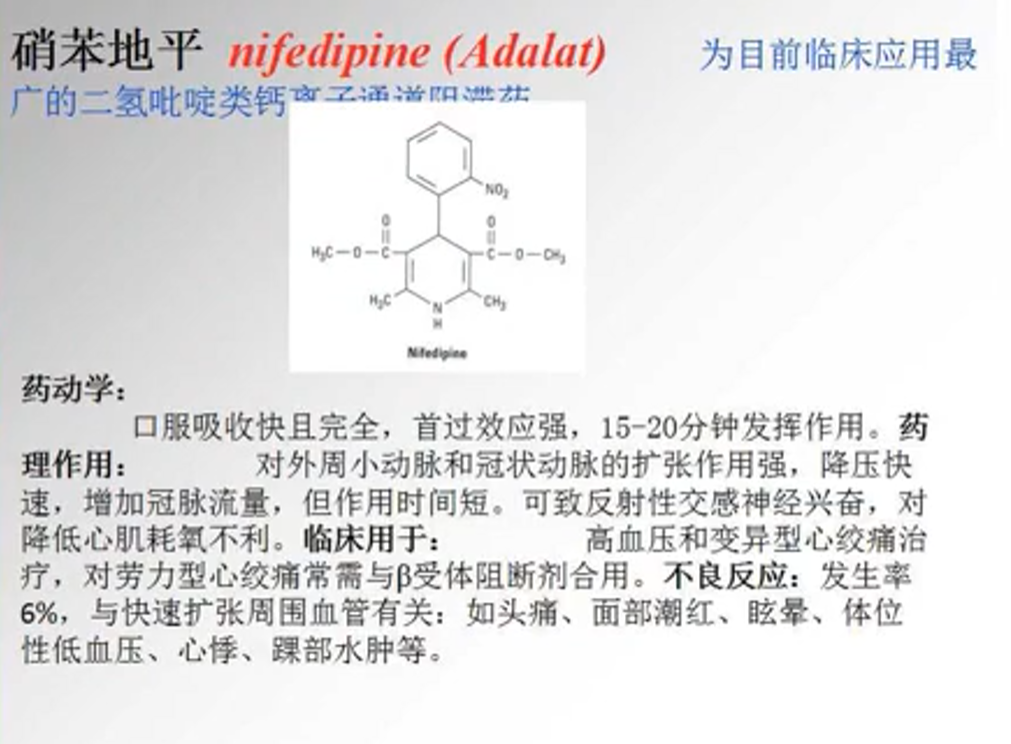
- 硝苯地平 nifedipine
- 维拉帕米 verapamil
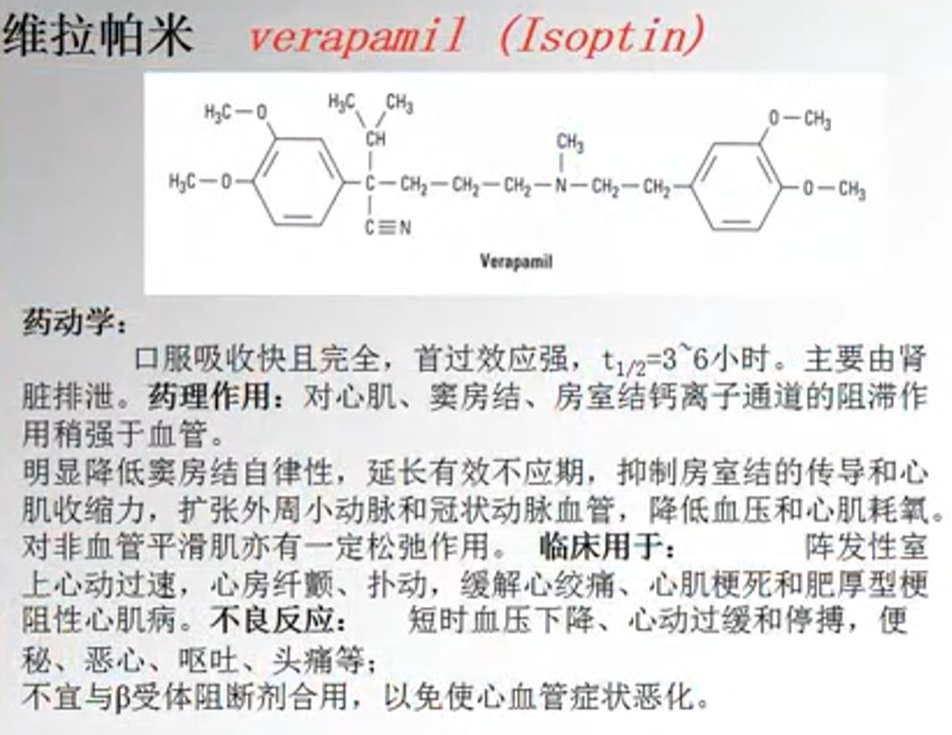
- 硝苯地平 nifedipine
- 作用机制
- 交感神经抑制药
- 分类

- β受体阻断剂作用机制
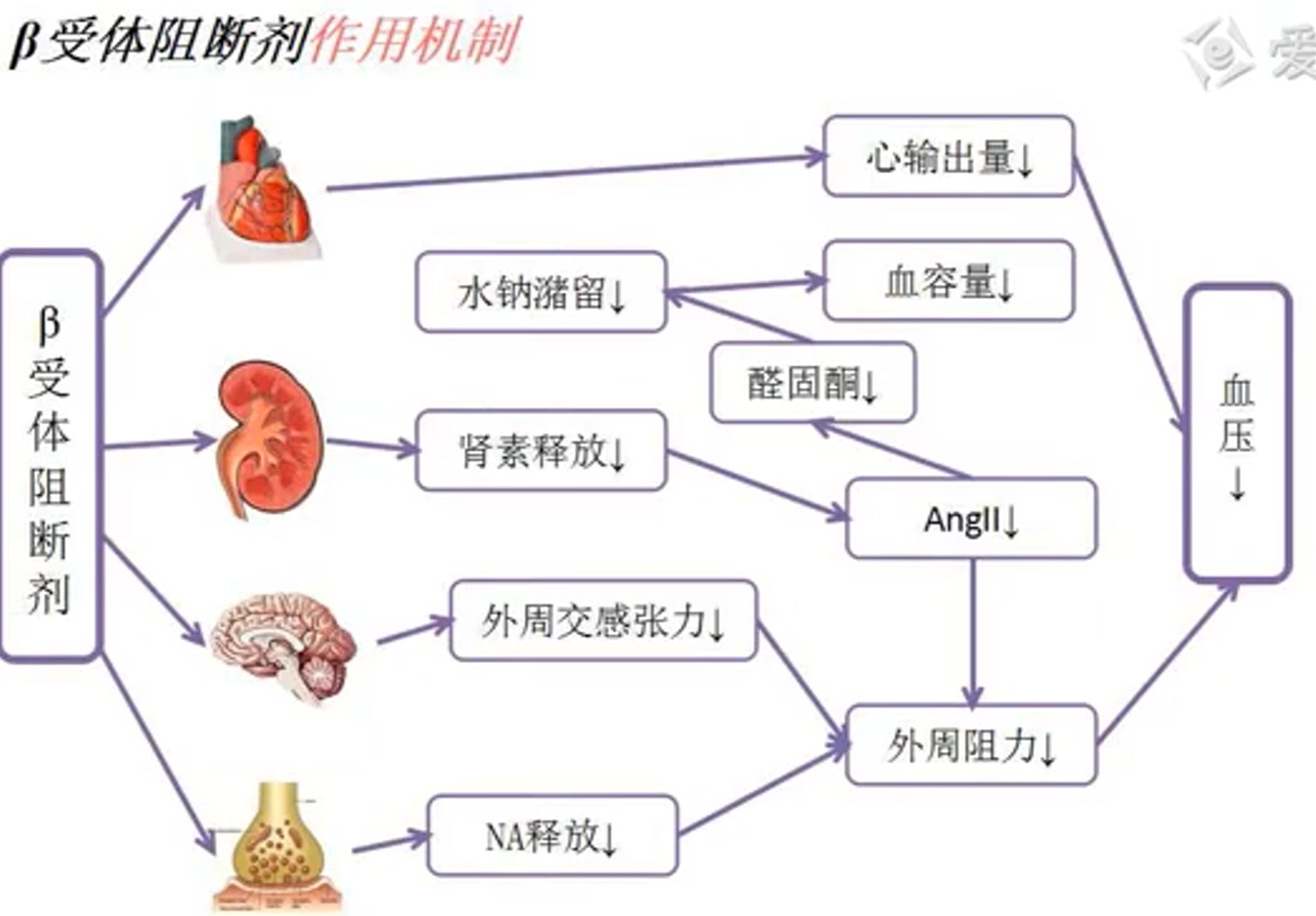
- 常见药物:普萘洛尔、卡维地洛(第三代)等,详见上一章
哮喘病人禁用
- 分类
- 血管扩张药
- 分类和机制

- 常见药物

- 分类和机制
- 利尿药
- 作用机制和应用

- 作用机制和应用
- 肾素-血管紧张素系统抑制药
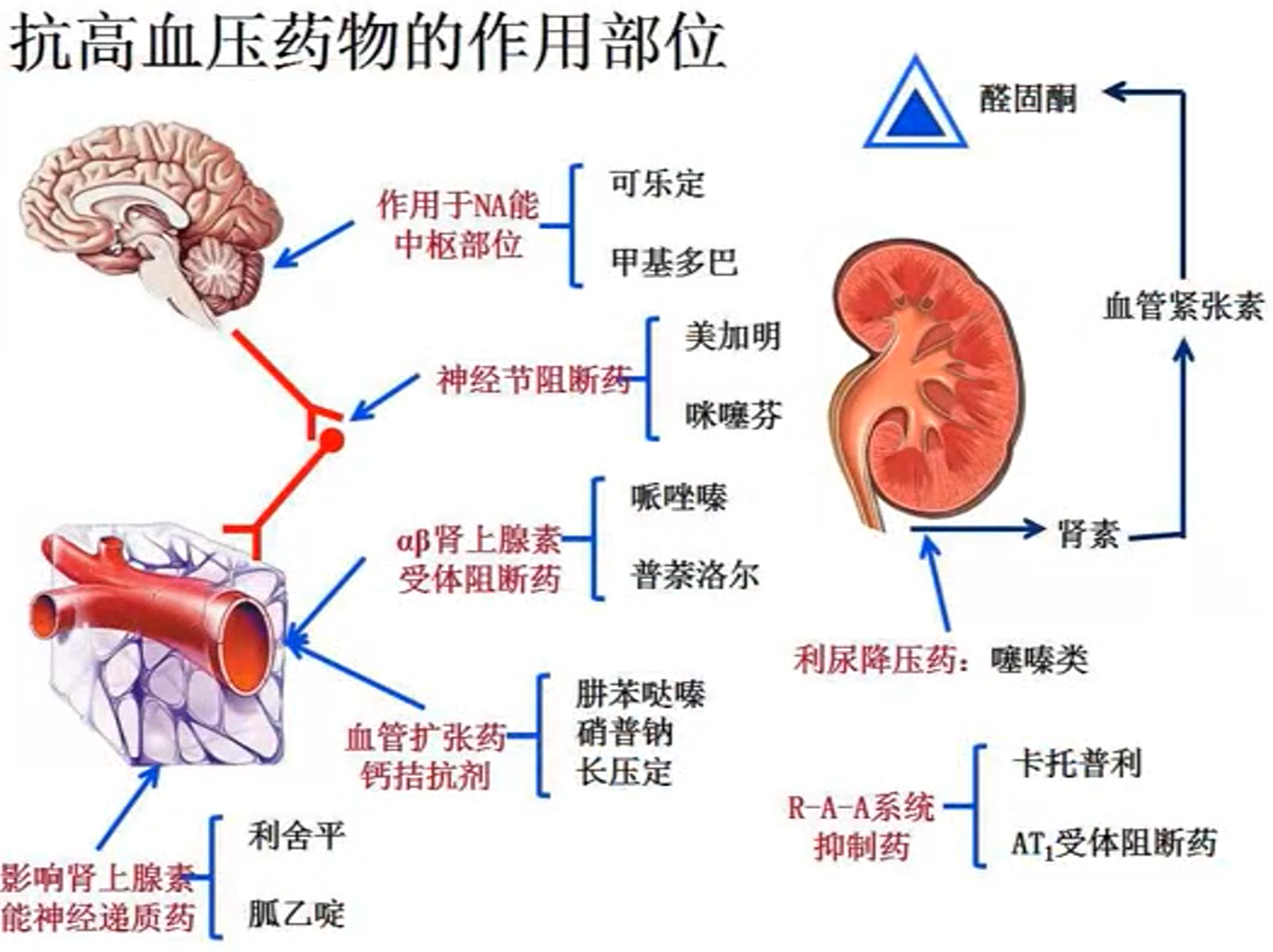
- 抗高血压药物的作用部位
抗炎症药理
炎症的生理病理基础
炎症的病理概述
- 定义:对损伤因子(生物性因子最常见)做出的以防御为主的反应
- 特征:
- 变质(可逆)/坏死(不可逆) -> 损伤(原因)
- 渗出 -> 抗损伤(也可能带来新的损伤)
- 增生 -> 抗损伤
- 中心-血管反应
- 血流动力学改变:血管收缩(很短) -> 血管扩张 -> 血流加速 -> 血流缓慢
- 理解血管扩张、血流加速的原因:抗损伤的过程需要血管内物质渗出,血流加快血管扩张可以加快渗出速度
- 理解血流缓慢:微血管通透性升高的结果。 富含蛋白质的液体向血管外渗出导致血管内红细胞浓集和粘稠度增加,最后扩张的小血管内挤满了红细胞,称为血流停滞(stasis)
- 血管通透性增加(液体蛋白渗出的主要原因,白细胞通过游走穿过内皮间隙,红细胞被动漏出血管)
- 炎症介质引起内皮细胞收缩间隙增大
- 内皮细胞胞吞胞吐作用增强
- 内皮细胞损伤,基底膜受损(烧伤、化脓菌、白细胞介导)
- 新生毛细血管高通透性
- 血流动力学改变:血管收缩(很短) -> 血管扩张 -> 血流加速 -> 血流缓慢
炎症的病理表现
- 血管扩张充血, 组织变红 - C5a, C3a, PG, P物质,组胺, 缓激肽, NO
- 炎症渗出物 -> 水肿
- 局部和全身发热 - PG, TNF, IL-1
- 疼痛 - 缓激肽, PG, P物质
- 功能障碍
- 血白细胞浓度
- 增高
- 多数细菌:中性粒细胞
- 寄生虫、过敏:嗜酸性粒细胞
- 某些病毒:淋巴细胞、单核细胞
- 下降
- 多数病毒:立克次体、原虫、伤寒杆菌
- 增高
不难发现PG前列腺素参与了大量炎症相关过程,因此使用非甾体抗炎药阻断前列腺素合成的酶COX1可以广泛地减轻炎症反应
炎症的病理类型(可略)
- 变质性炎(坏死) - 中毒性心肌炎、病毒性肝炎、乙脑、阿米巴
- 渗出性炎
- 浆液性炎:黏膜(感冒)、浆膜(浆膜腔积液)、滑膜(风湿性关节炎)、皮肤(水疱)
- 纤维素/纤维蛋白性炎
- 黏膜/假膜性炎:细菌性痢疾、白喉
- 浆膜:绒毛心
- 肺:大叶性肺炎(主要是肺炎链球菌肺炎)
- 化脓性炎(中性粒细胞)
- 举例:小叶性肺炎、流脑、肾盂肾炎、急性阑尾炎、淋病
- 类型:表面化脓和积脓、蜂窝织炎
- 出血性炎
- 增生性炎
炎症反应的理解
解热镇痛抗炎药
总论
-
定义: 解热镇痛抗炎药(antipyretic-analgesic and anti-inflammatory drugs)= 非甾体抗炎药(non-steroidal anti-inflammatory drugs,NSAIDs)
-
临床效果:解热镇痛抗炎抗风湿
-
总体作用机制:抑制环氧酶(COX)的活性,使得机体内的前列腺素(PG)水平降低。由于前列腺素是导致发热、疼痛、炎症等反应的关键炎性介质,引起降低其表达水平可以达到目标效果
-
扩展定义:凡是能起降低前列腺素水平的药物,最终效果都可以解热镇痛抗炎
-
分类(甾核:steroid nucleus)
- 甾体类抗炎药(化学结构存在甾核)
- 糖皮质激素类似物,抑制减少磷脂酶A2(PLA2),减少花生四烯酸的产生进而降低COX效能,减少糖皮质激素的产生。
- 非甾体类抗炎药
- 有机酸类化合物,不具有甾体结构,最终效果还是降低COX效能,减少糖皮质激素产生
- 甾体类抗炎药(化学结构存在甾核)
-
宏观理解
- 正常机体内细胞膜上存在膜磷脂,组成细胞膜,但不参与炎症反应过程
- 机体内有感染或者其他损伤性因素时,这些因素会刺激增强体内的磷脂酶A2(PLA2)的水平和效能。
- PLA2催化膜磷脂转化为花生四烯酸(AA)。而花生四烯酸是一种中间产物,它随后会在被不同的酶系催化而转变为不同的物质,从而产生对应的生理或病理效果。另外也会产生血小板活化因子(PAF)
- 花生四烯酸在不同的酶系的催化作用下转化为其他的自体活性物质
- 在脂氧酶(LO)作用下转换为白三烯(leukotriene,LT)脂氧素(lipoxin)和羟基环氧素(hepoxilin,HX)
+ 这三个物质都是免疫应答中的重要细胞因子,都具有促使炎症和免疫细胞趋化的作用,让大量的免疫细胞趋化到发生炎症的局部,从而释放炎症介质,进而导致免疫系统反映,并起到收缩支气管,增加血管通透性等。 > 但是NSAIDs药物对LO没有作用,主要是糖皮质激素抑制了PLA2- 环氧化酶催化下转化为“环内过氧化物”,不稳定会发生进一步转化
- 合成多种前列腺素(PGF2, PGD2, PGE2),并且促进产生炎症、发热、致痛效果
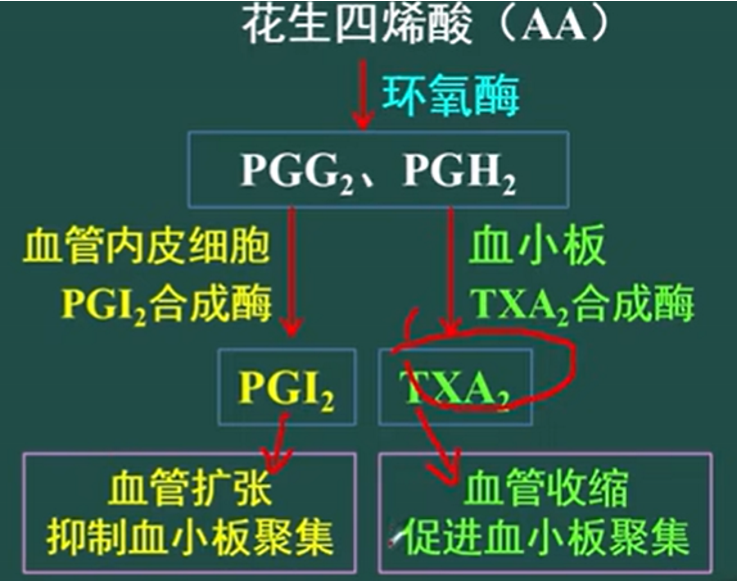
- 前列腺素合成酶(PGIS)催化合成前列腺素I2(PGI2),主要作用为抗凝血,使得血小板解聚和舒张血管的作用
- 血栓素合成酶(thromboxane synthetase)催化下转变为血栓素A2(TXA2,thromboxane A2):血栓素A2的作用是收缩血管,并且促进血小板聚集(Platelet polymerization)。
由上述机制可以理解,使用NSAIDs药物的时候会由于血栓素A2水平下降,使得血小板凝聚力下降;同时由于PGI2是促使血管舒张,现在PGI2减少所以血管收缩,引起血压升高
- 能作用于以上机制的药物
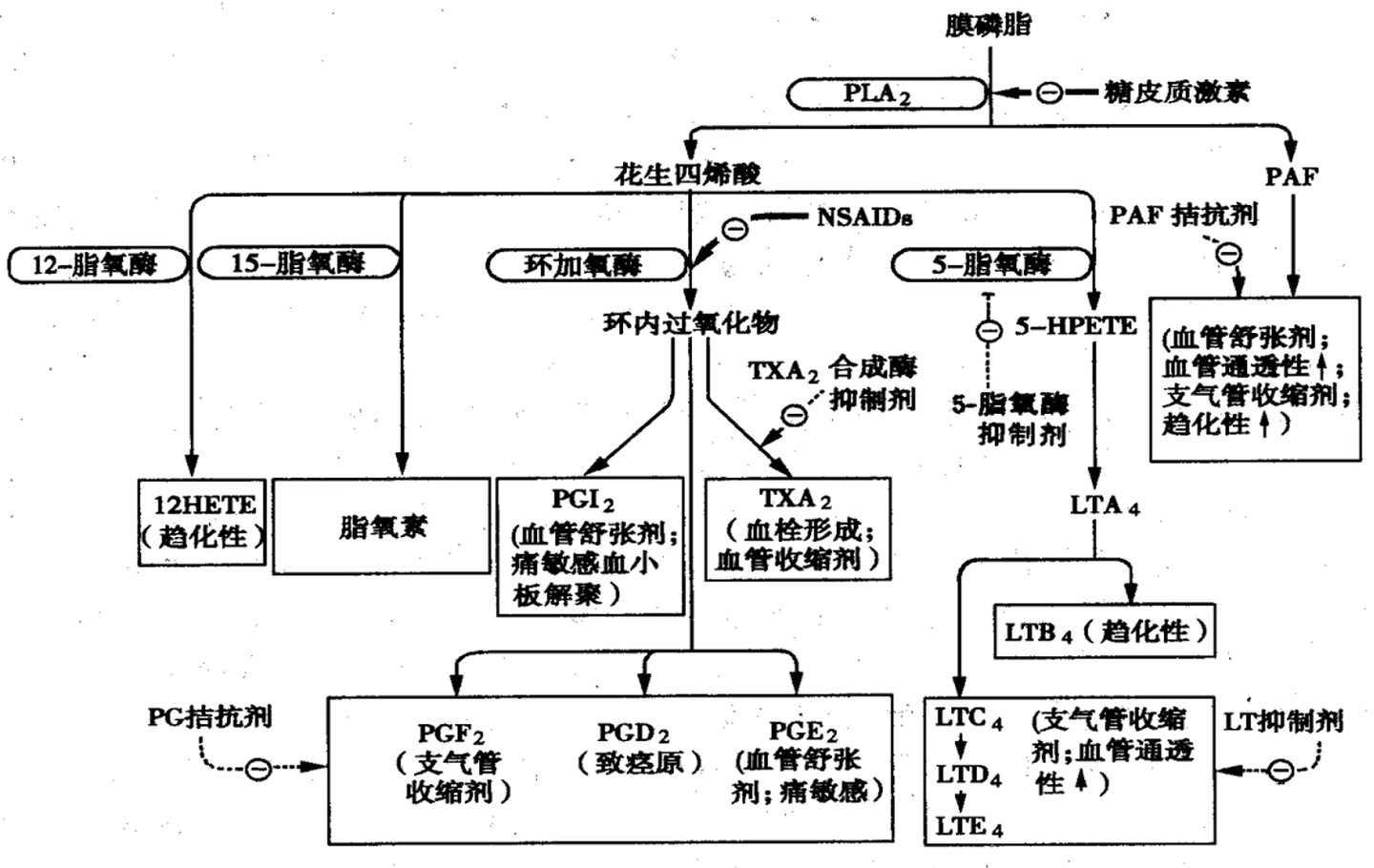
- 糖皮质激素=甾体类抗炎药:作用于磷脂酶A2,降低花生四烯酸及其代谢产物的水平
- 非甾体类抗炎药(NSAIDs):作用于环氧酶(COX),使得花生四烯酸的转化为前列腺素类似物的代谢通路被抑制
- 血栓素A2合成酶抑制剂:降低血栓素A2,降低血小板的聚集作用,起抗凝血效果(PGI2本身同理,即“依前列醇(epoprostenol))
- 白三烯受体拮抗药:白三烯是花生四烯酸在脂氧酶的转化产物,会趋化炎症细胞加重炎症反应。但是NSAIDs不能抑制脂氧酶,不能降低表达水平;然而可以使用糖皮质激素作用于代谢上游
-
NSAIDs类药物的具体药理效果和机制
- 解热镇痛抗炎效果
- 解热作用
- 机制:通过抑制COX酶,减少促使体温异常升高的物质(前列腺素E2),不直接作用于下丘脑体温调节中枢。
- 效果:减少体温因为炎症或组织损伤引起的异常升高
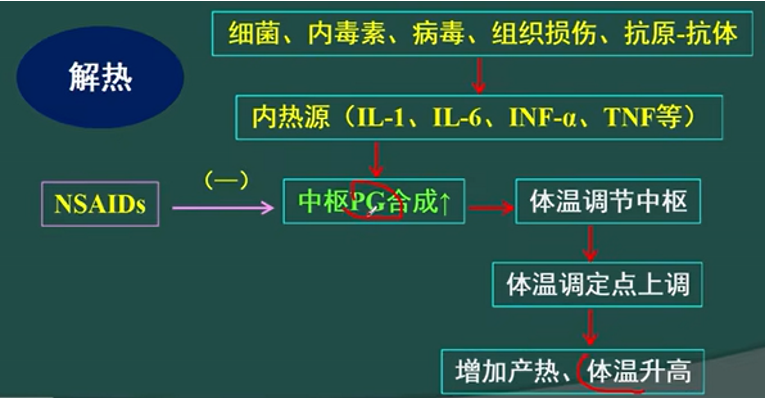
- 镇痛作用
- 机制:NSAIDs药物起到的镇痛效果是“外周性”的,其靶点并不是中枢神经系统。前列腺素所影响的是“第一级传入(感觉)神经元末梢对痛觉信息的感知”,而不影响痛觉信号在上行通路中的传导(这其实是前面阿片类镇痛药的靶点),也不影响大脑皮层根据传入的痛觉信号最终产生痛觉感受(这其实是全麻药的靶点)。
痛觉的产生有两种类型:一种是伤害性因素直接作用于传入神经元的末梢产生动作电位,第二种是伤害性因素引起对应区域的炎性反应,炎症因子刺激传入神经的神经末梢
PG会起痛觉感知增敏的效果,因此NSAIDs只能减少第二种疼痛感,但对第一种的直接刺激无效- 效果:NSAIDs药物对急性锐痛和严重的创伤性剧痛无效(因为他们是“创伤因素直接刺激感觉神经末梢而导致的疼痛”),但是对炎症和慢性组织损伤引起的疼痛有效。
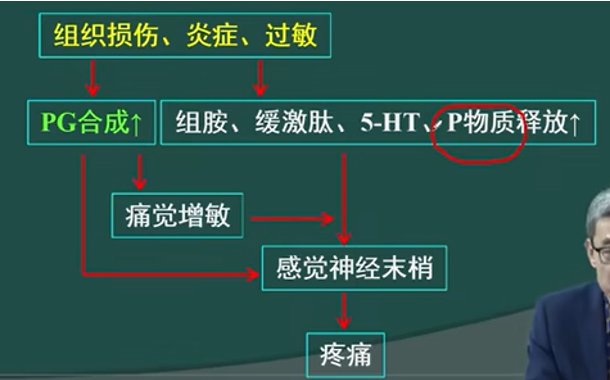
- 抗炎作用
除了对乙酰氨基酚(苯胺类NSAIDs)之外,其他的NSAID仍然都是有抗炎作用的,因为对乙酰氨基酚对外周的“抑制PG生成”的能力很差,并不能缓解炎症部位的反应。
- 机制:当机体受到非特异性致炎物质或机制的影响时,其会释放出更多“前列腺素(PG)”和“缓激肽(bradykinin)”,这两个物质有促进血管扩张、血管通透性增加的效果,进而引起组织红肿疼痛。NSAIDs药物通过抑制COX酶,降低外周和中枢的PG生成以此达到抗炎效果
- 效果:降低受到损伤性刺激的部位处前列腺素类物质的分泌水平,一定程度阻断“红肿热痛”的机制
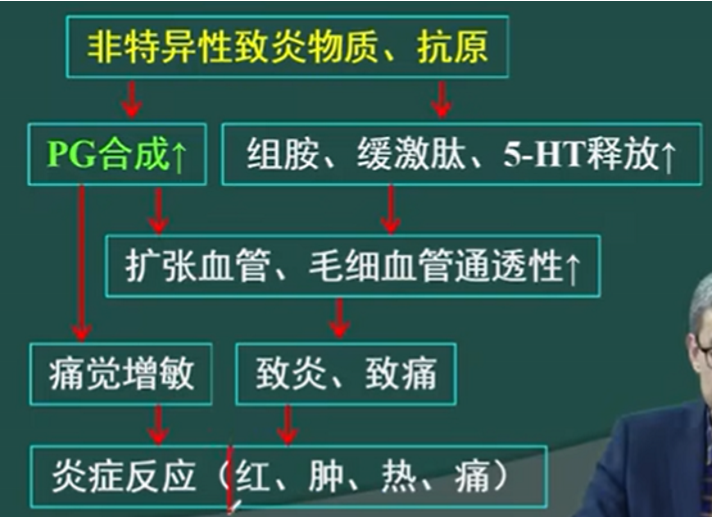
- 解热作用
应该明确的是,NSAIDs类药物其实只是有效减轻了机体的炎症反应,减少了患者的不适体验,但是治标不治本,没有从根本解决引起炎症的始动因素。
- 解热镇痛抗炎效果
-
对COX-1和COX-2的区别和理解
- COX-1负责产生“维持机体正常生理过程所需的前列腺素类物质或血栓素等”从而对胃粘膜、血管、血小板、肾脏血流灌注等起到良性促进作用。
- COX-2在正常情况下仅表达于机体内的小部分部位,但是在机体受到损伤性因素的情况下,机体发生炎症反应的时候,COX-2会在全身各处增加表达,从而在全身各处起到致炎、致热、致痛的效果。
- 如果抑制了COX-2的表达可以气道解热镇痛抗炎的效果,但是如果同时抑制了COX-1的水平就会引起各种副作用
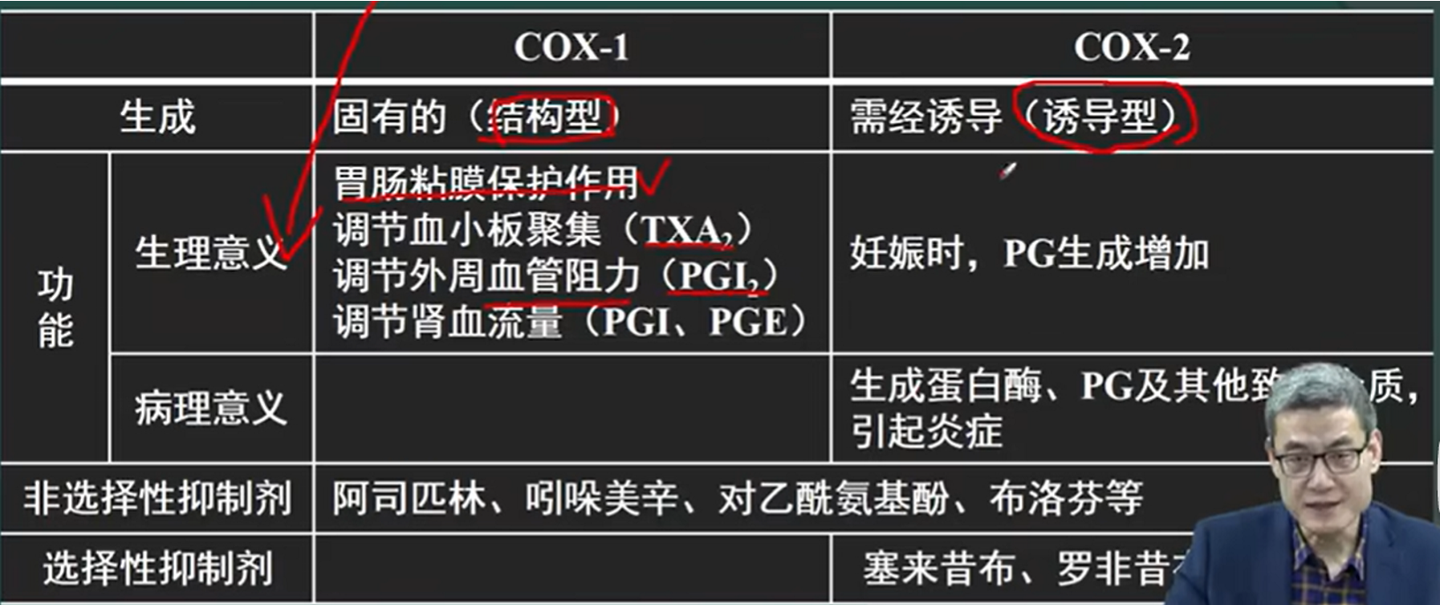
-
NSAIDs药物常见副作用
- 胃肠道出血或溃疡:COX-1的作用下,在胃肠道处粘膜产生前列腺素类物质,有助于保护胃肠道粘膜的功能,抑制胃酸分泌(PGI2可以抑制胃酸分泌;PGE2和PGF-2α可以刺激胃肠道粘膜生成更多保护性的黏液),非选择性NSAIDs阻断COX-1,引起胃黏膜的损伤进而引起出血
- 肾脏损伤:在肾动脉处,会在COX-1的作用下生成PGE2和PGI2,这些物质会在一定程度上扩张肾血管,从而将肾脏血流灌注维持在较高水平。非选择性NSAIDs阻断COX-1,造成肾动脉的扩张程度下降,肾脏的血流灌注会减少,如果本身就有血管收缩效应的患者,可能引起急性肾脏衰竭
- 血小板聚集障碍:身体会自发产生活性物质,其中PGI2和TXA2相互拮抗。PGI2主要在血管内皮细胞处由血管内皮细胞产生的COX-1催化而来,其效果是促使血管扩张,并且抑制血小板聚集;TXA2主要在血小板处由血小板中的COX-1催化而来,其作用是促使血管收缩,并且促进血小板聚集。PGI2主要在血管内皮细胞处产生,而血管内皮细胞是成熟的有功能的细胞,它是有能力随着时间推移重新合成一批新的有功能的COX-1。TXA2,其主要在血小板中形成,但是成熟的血小板是没有细胞核的细胞,它寿命短,是不具有新产生一批COX-1的能力的。用药阿司匹林经过一段时间后,抑制血小板凝集的PGI2的回升速度会显著快于促进血小板凝集的TXA2的回升速度,所以,此时血小板凝集被抑制的程度会越来越大,从而患者的止血和凝血机制的活动力会越来越低,可以导致显著的出血倾向
常见的NSAIDs药物
-
阿司匹林(aspirin)=乙酰水杨酸(acetylsalicylic acid)
- 药代动力学特点
- 乙酰水杨酸在体内会经过代谢,最终以水杨酸的结构生效,直接服用会对胃肠道刺激过高
- 阿司匹林是弱酸性物质,在酸性环境下更容易被吸收。
磷脂双分子层倾向于阻碍带电和亲水的分子,通过亲脂不带电荷的分子,所以分子状态的时候更容易跨膜,离子状态倾向于留在特定部位
- 如果出现阿司匹林苯巴比妥这类弱酸性药物中毒的情况,应该采取碱化尿液的方法(即静脉滴注碳酸氢钠),因为尿液酸性较强的时候会促进肾小管对阿司匹林的重吸收,碱性的时候会抑制,加快排泄解毒
- 药理作用
- 解热镇痛抗炎
- 抗血小板聚集和抗血栓生成
- 机制:小剂量破坏血小板的COX-1,因为血小板是细胞碎片不具备重新生成COX-1的能力,以此来降低TXA2的表达,让PGI2占主导来抑制凝血;大剂量可能引起PGI2合成也受到抑制反而药效消失
- 副作用
- 主要都是由COX-1受抑制导致
- 胃肠道反应: 恶心呕吐胃肠道不适,可能胃出血
- 血小板凝血障碍
- 常规过敏反应
- 药代动力学特点
-
其他非选择性NSAIDs
- 苯胺类(Aniline)
- 对乙酰氨基酚(acetaminophen)
- 特点:基本不具有抗炎和抗风湿作用,只有解热镇痛作用,因为对外周COX作用较小
- 过量可能引起肝衰竭
- 对乙酰氨基酚(acetaminophen)
- 芳基丙酸类:代表药物是布洛芬(ibuprofen)、萘普生(naproxen)、非诺洛芬(fenoprofen)、酮洛芬(ketoprofen)、奥沙普秦(oxaprozin)等
- 吲哚类:吲哚美辛(indomethacin)
- 苯胺类(Aniline)
-
选择性NSAIDs————选择性抑制COX-2
- 只抑制COX-2而不抑制COX-1能够很大程度上减少副作用
- 本类药物的代表要是三个:塞来昔布(celecoxib)、罗非昔布(rofecoxib)和尼美舒利(nimesulide)、美洛昔康(meloxicam)
- 但是由于应用发现,这类药物的使用会导致心脑血管事件的发生率升高,其中的罗非昔布甚至已经因为这个原因停止生产