
Bioinformatics study notes
Common sence on bioinformatics
差异分析
- 为什么需要做差异分析?
- 差异分析就是分析两组数据在何种指标下有差异。在转录组的基因差异表达分析中,一般的筛选标准是基因表达差异倍数大于2、并且FDR≤0.05为显著差异的基因,当然这个标准也可以根据实际数据调整。
- 差异分析原理
- 差异分析的原理就是判断组间(处理)差异是否显著大于组内(误差)差异。
由于二代数据得到的基因表达丰度是reads count,属于离散型分布,所以主流的方法是用负二项分布(属于离散型分布)模型进行检验。主流差异分析软件edgeR、Deseq都采用负二项分布。二代数据工具不能使用t检验、方差分析等这些基于正态分布模型的检验方法。
- 分析步骤
- 数据准备,提供符合格式的数据表格
- 参数设置(检验值、检验阈值、差异倍数、离散系数)
- 解读结果
- 差异比较结果柱状图
- 差异基因火山图
- 差异分析数据要求
- 表达矩阵和分组信息(count矩阵)
富集分析
- 为什么需要做富集分析?
- 在我们进行差异表达分析的时候,我们会得到很多的差异表达基因。这些基因如果只是按照基因名放到哪里的话,我们很难找到一个规律说这些有基因之间有什么关系,所以为了更情况的看清楚这些基因的功能,我们就使用了富集分析。
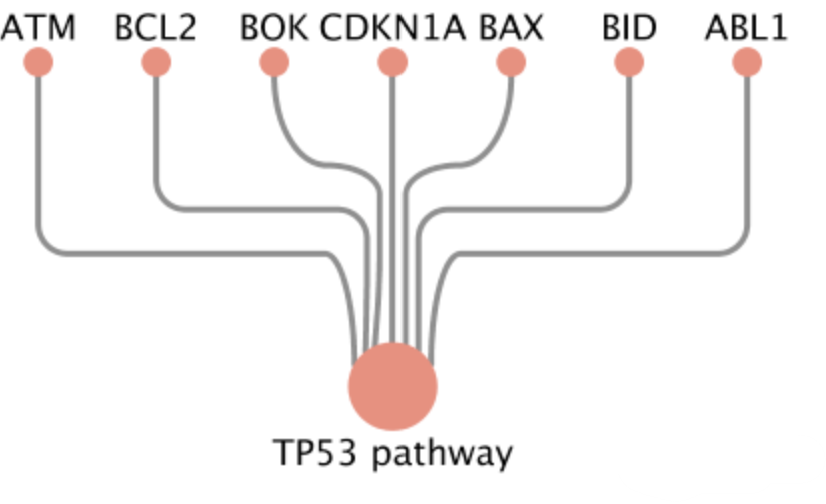
- 利用富集分析,我们就可以把很多看着杂乱的差异基因总结出一个比较整体反应事件发生的概述性的句子。例如:TP53信号通路和胃癌的发生有关。而不是说BAX、BID、ABL1、ATM、BCL2、BOK、CDKN1A这7个基因和胃癌的发生有关系。
- 富集分析数据要求
- GO/KEGG:两种分析基因和通路的数据库,输入数据均是差异表达分析的结果,数据内容为差异基因名称(编号)
- GSEA:一种不依赖于差异表达分析的算法(也有自己的细分数据库MSigDB),数据类型为差异基因名称和Log2FoldChange(差异倍数的对数值)
- 如何解读分析结果
- 差异分析:
+ - 富集分析:
- GSEA:
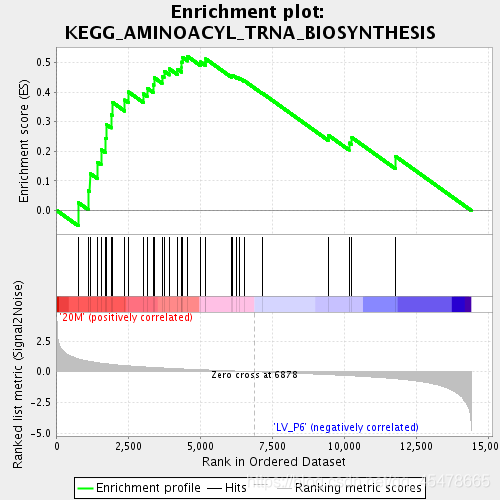
第一部分:为基因 Enrichment Score 的折线图,横轴为该基因下的每个基因,纵轴为对应的 Running ES,在折线图中有个峰值,该峰值就是这个基因集的 Enrichemnt score,峰值之前的基因就是该基因集下的核心基因,核心基因就是在单细胞很重要很关键的基因,如果基因在顶部富集,我们可以说,从总体上看,该基因集是上调趋势,反之,如果在底部富集,则是下调趋势;
第二部分:也就是hit值,也就是图中的黑线出现的位置,这个黑线可以看出第一部分每次折现点的基因的位置;
第三部分:这是rank值图,采用了 Signal2Noise 算法,我对他的理解是,纵轴它反映了基因集中的每个基因的相对表达量,横轴可以看出这是这个基因在基因数据集中出现的位置。
- GSEA:
- 差异分析:
Common bioinformatics analysis methods
KEGG/Go analysis
-
What is KEGG?
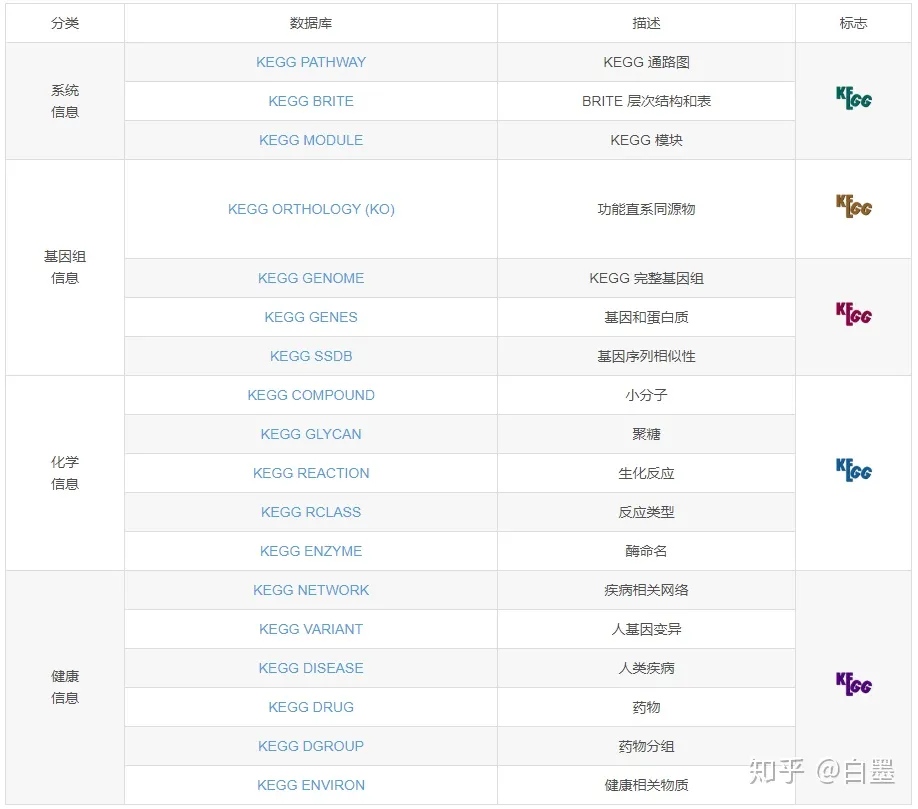
Kanehisa实验室 在1995年开发的KEGG数据库,全称为 Kyoto Encyclopedia of Genes and Genomes(京都基因与基因组百科全书)。它拥有多个子数据库,包含基因组,生化反应,生化物质,疾病与药物,以及 最常用PATHWAY通路信息 。
KEGG Links什么是富集分析:这里pathway富集的含义与GO富集的含义相同,也是表示差异基因中注释到某个代谢通路的基因数目在所有差异基因中的比例显著大于背景基因中注释到某个代谢通路的基因数目在所有背景基因中的比例。
-
What is GO?
GO数据库,全称是Gene Ontology(基因本体),他们把基因本身的功能分成了三个部分分别是:细胞组分(cellular component, CC)、分子功能(molecular function, MF)、生物过程(biological process, BP)Difference with KEGG? KEGG是通路相关的数据库,GO是基因自身功能的数据库。
Relationship? GO和KEGG是两个数据库,里面有每个基因相关的功能信息,而富集分析就是一个把这些功能进行进行整合计算的算法 -
Database Structure
-
KEGG pathway
每个通路都由一个五位数字标识,后跟以下任意一个:map,ko,ec,rn和三字母或四字母生物代码,它们分别代表五种通路类型:- map编号:代表reference pathway,根据已有的知识绘制的、概括的、详尽的具有一般参考意义的代谢图。 一个点同时表示一个基因,这个基因编码的酶或这个酶参加的反应
- org编号:物种特异性通路,这里就是将K编号基因(直系同源基因,后面会介绍)换为每个物种中对应的基因
- ko编号:KO通路中的点表示直系同源基因
- ec编号:EC通路中的点表示相关的酶
- rn编号:化学反应通路中的点只表示该点参与的某个反应、反应物及反应类型
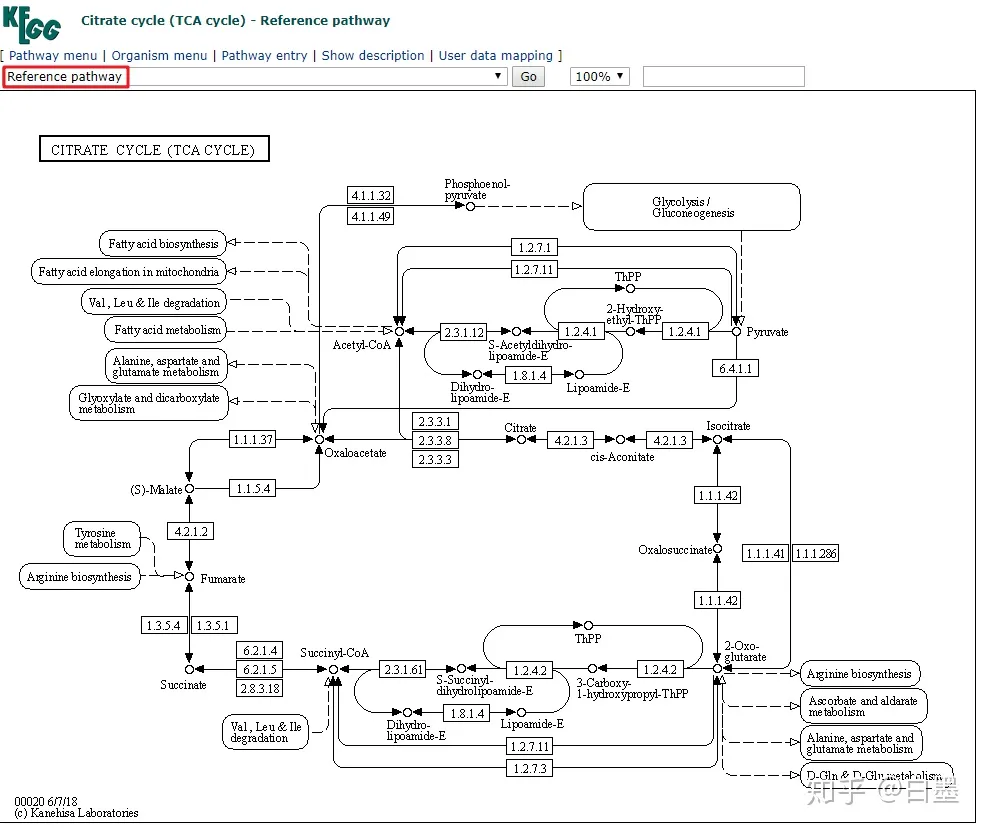
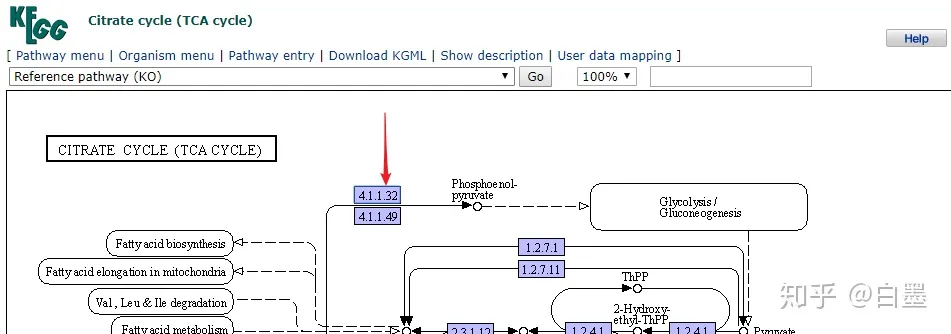
1). Reference pathway
这里以 TCA循环 的通路图为例,进入参考通路图(Reference pathway)。
2). 物种特异性通路 (org)
我们选择人的物种名
homo sapiens(human),点击Go。可以看到与Reference pathway 图(map00020)不同的是有物种特异性基因被标注为绿色,而且通路编号为hsa00020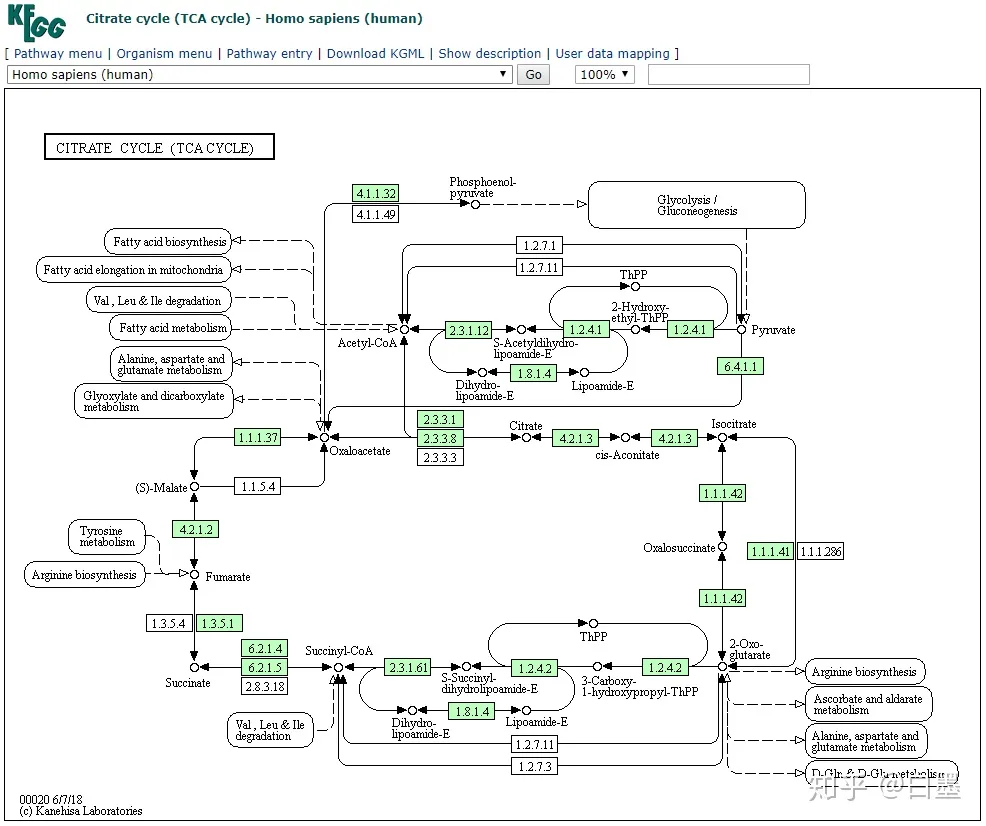
点击绿色基因,会进入Gene详细信息
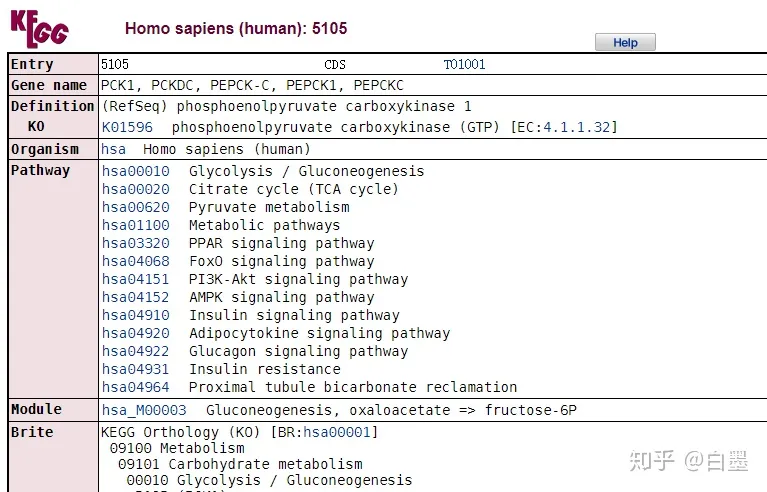
3). 直系同源物通路 (ko)
KEGG的开发者根据不同生物之间基因和基因组的保守和变异,引入直系同源物(KO)的概念。
蓝色框超链接到从原始版本中选择的KO条目
进入PCK的直系同源基因信息
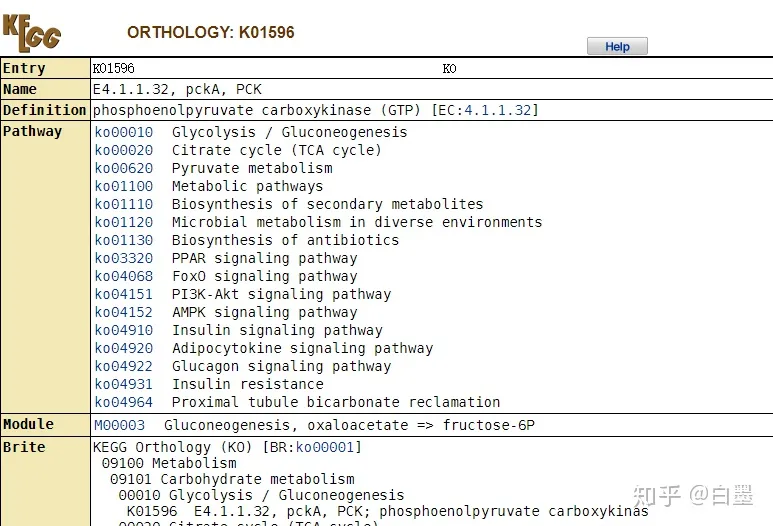
4). 酶通路(ec)
蓝色框超链接到从原始版本中选择的ENZYME条目
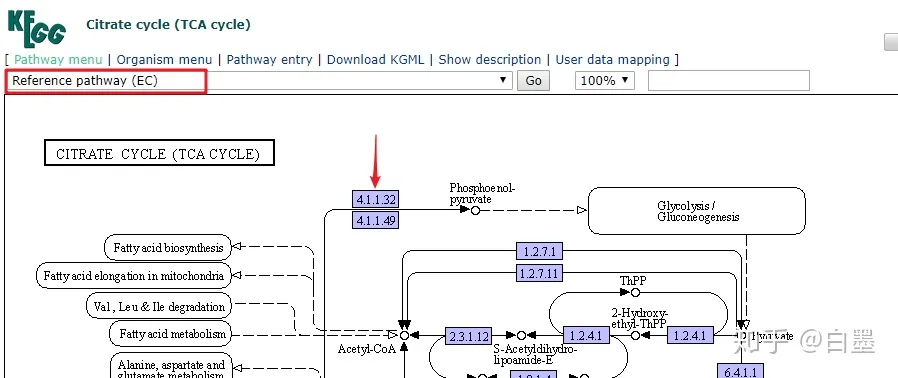
进入ENZYME
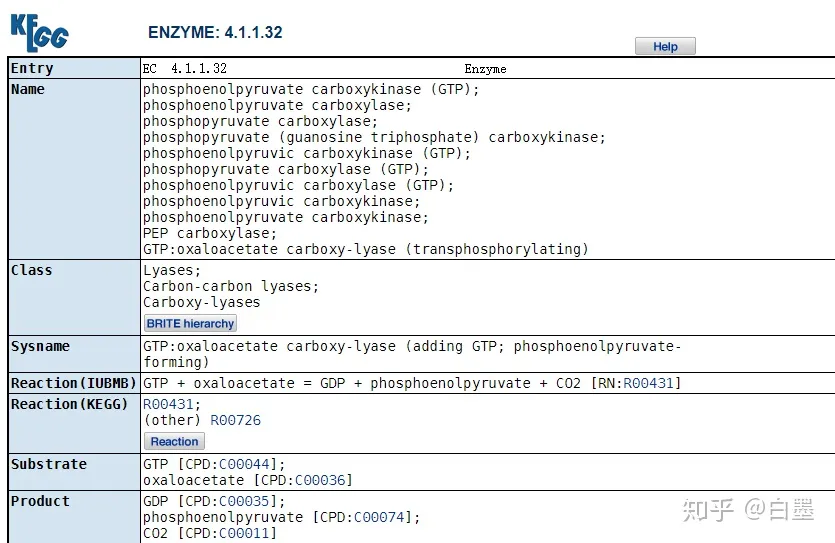
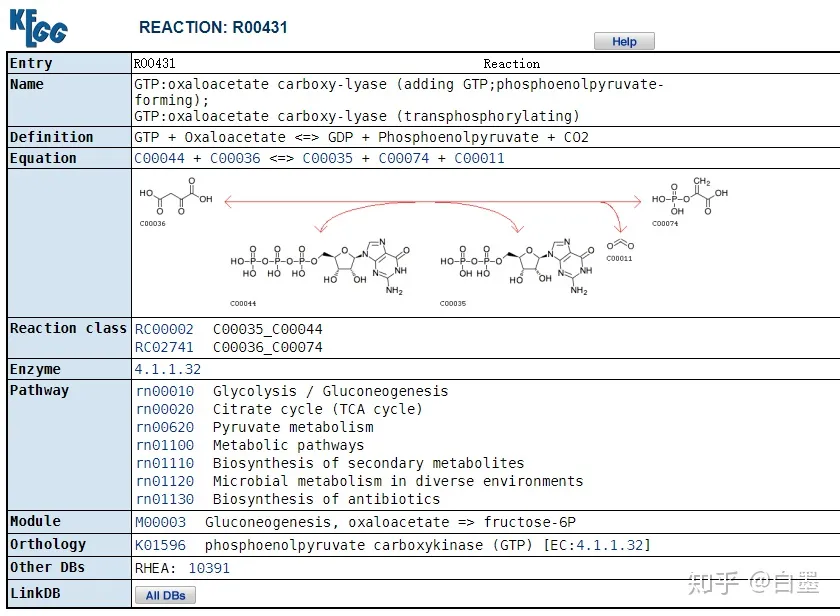
5). 反应通路 (reaction)
反应通路 (reaction)
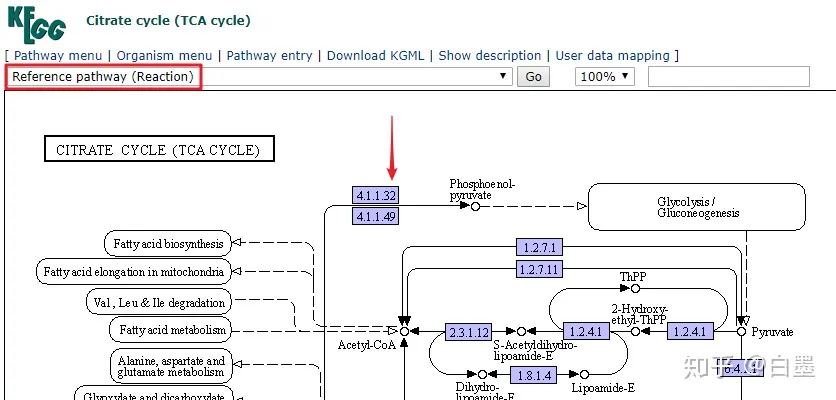
点击后进入对应的反应信息界面,如下图
-
GO Database
-
kegg/GO pipeline
- 载入差异化表达数据并调整数据格式
1
2
3
4
5
6
7
8
9#载入差异表达数据,只需基因ID(GO,KEGG,GSEA需要)和Log2FoldChange(GSEA需要)即可
info <- read.csv("./DEGs_test_data/Del19_to_p.L858R_raw.csv")
#指定富集分析的物种库
GO_database <- 'org.Hs.eg.db' #GO分析指定物种,物种缩写索引表详见http://bioconductor.org/packages/release/BiocViews.html#___OrgDb
KEGG_database <- 'hsa' #KEGG分析指定物种,物种缩写索引表详见http://www.genome.jp/kegg/catalog/org_list.html
#gene ID转换
gene <- bitr(info$gene,fromType = 'SYMBOL',toType = 'ENTREZID',OrgDb = GO_database)- 带入R包分析
1
2
3
4
5
6
7
8
9
10
11
12GO<-enrichGO( gene$ENTREZID,#GO富集分析
OrgDb = GO_database,
keyType = "ENTREZID",#设定读取的gene ID类型
ont = "ALL",#(ont为ALL因此包括Biological Process,Cellular Component,Mollecular Function三部分)
pvalueCutoff = 0.05,#设定p值阈值
qvalueCutoff = 0.05,#设定q值阈值
readable = T)
KEGG<-enrichKEGG(gene$ENTREZID,#KEGG富集分析
organism = KEGG_database,
pvalueCutoff = 0.05,
qvalueCutoff = 0.05)
GSEA analysis
-
What is GSEA?
基因集富集分析(Gene Set Enrichment Analysis,GSEA):用一个预先定义的基因集中的基因来评估在与表型相关度排序的基因表中的分布趋势,从而判断其对表型的贡献。
-
GSEA analysis principle
-
背景基因排序:将全部基因按照某种指标(差异分析p值,表型相关性,表达量等)进行排序,比如log2FC排序。
-
目标基因富集:将某个特定类型的基因在排序表中标出,目标基因可以是某个通路或GO terms的基因等。
-
计算富集分数:使用加权法,计算ES值变化。对位于中部(与性状相关性低)的部分采用较小的权值,所以越集中在两端,与表型的相关性越高。ES曲线最大值为富集分数(Enrichment Score)。
-
Permutation test:对基因集的ES值进行显著性检验及多重假设检验,从而计算出显著富集的基因集。
-
- 问题:一个富集到的通路下,既有上调差异基因,也有下调差异基因,那么这条通路总体的表现形式究竟是怎样呢,是被抑制还是激活?
- 解决方案:GSEA的输入是一个基因表达量矩阵,其中的样本分成了A和B两组,首先对所有基因进行排序,用来表示基因在两组间表达量的变化趋势。
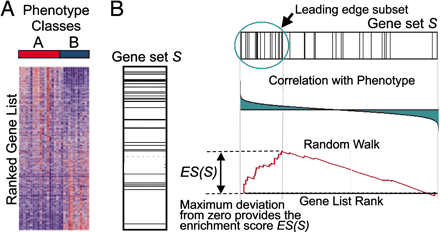
GSEA分析的是一个基因集下的所有基因是否在这个排序列表的顶部或者底部富集,如果在顶部富集,我们可以说,从总体上看,该基因集是上调趋势,反之,如果在底部富集,则是下调趋势。
- GSEA pipeline
Proteomics analysis
- Proteomics pipline
- 原始谱图搜库: 得到大量蛋白质的鉴定和定量信息
- 定量数据统计和展示: 关注蛋白鉴定数目、肽段鉴定数目、肽段分布情况以及样品相关性和重复性
- 差异表达分析: 找出不同处理组间差异表达蛋白
- 功能注释和富集分析: 对蛋白的功能、分布、参与的通路等方面进行注释和富集,为后续生物学和功能机制的研究提供参考和线索
- 工具:GO/KEGG
- 互作网络分析: 寻找蛋白质之间的关联性
- 工具:STRING
具体操作步骤(以赛默飞下机数据.raw为例)
- Step1: .raw质谱原始数据处理
- 使用ProteoWizard MSConvert工具将.raw转换为开放格式.mzXML/.mzML
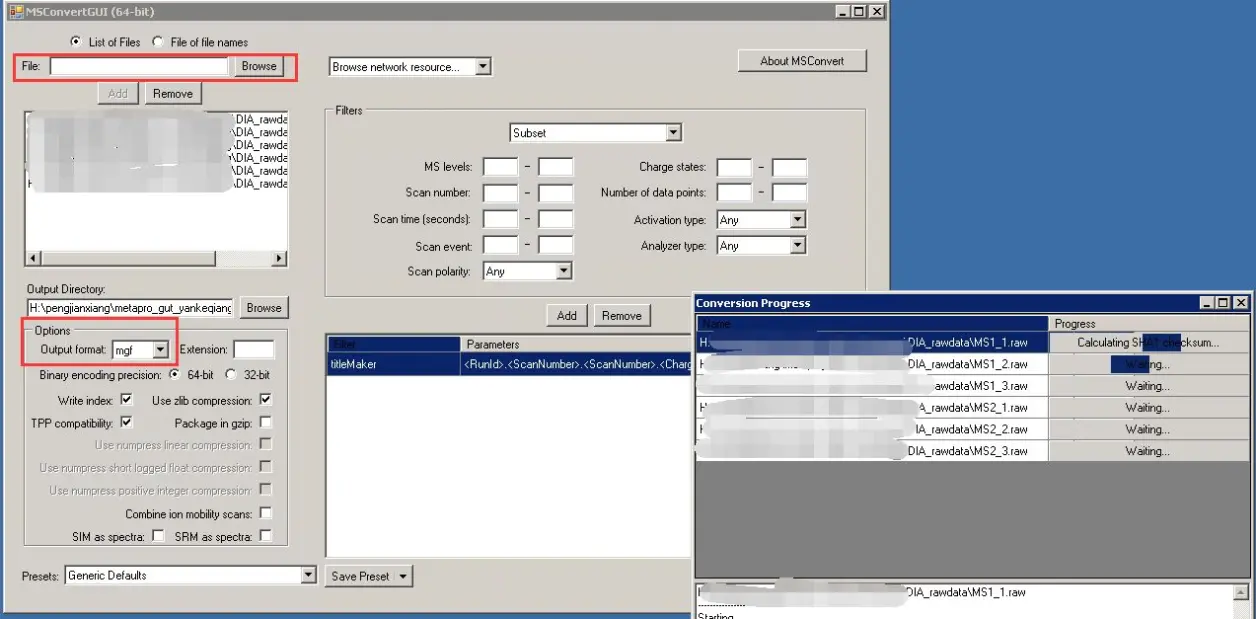
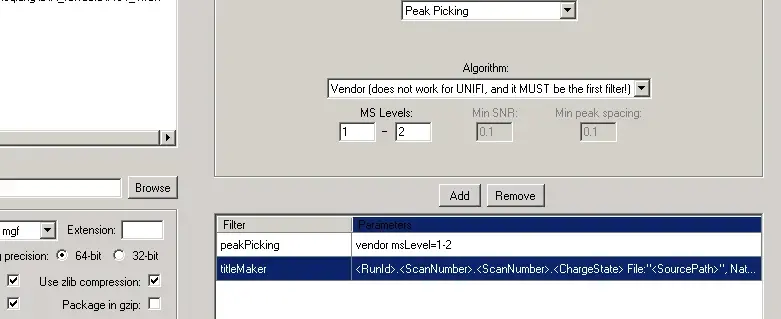
使用默认参数不过滤的话,转换的文件会非常大(比源文件增加数倍,因为它把一些无强度的峰也加进去了),速度也巨慢。一般Filters需要设置为Peak Picking,MS Levels设为1-2。
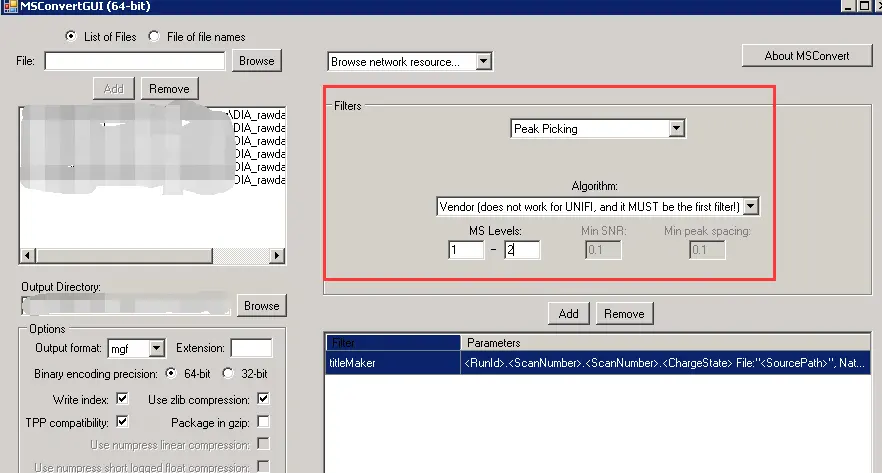
而且要注意,选择后记得按Add,否则还是没有加入peakPicking,并且要将peakPicking放在第一行(行可拖动)。
- 使用ProteoWizard MSConvert工具将.raw转换为开放格式.mzXML/.mzML
- Step2: